 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4xwy: Crystal structure of human sepiapterin reductase in complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xwy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human sepiapterin reductase in complex with an N-acetylserotinin analogue | ||||||
 Components Components | Sepiapterin reductase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / inhibitor / complex | ||||||
| Function / homology |  Function and homology information Function and homology informationsepiapterin reductase (L-erythro-7,8-dihydrobiopterin forming) / sepiapterin reductase activity / aldo-keto reductase (NADPH) activity / tetrahydrobiopterin biosynthetic process / eNOS activation / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / nitric oxide biosynthetic process / NADP binding / extracellular exosome / nucleoplasm / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.35 Å | ||||||
 Authors Authors | Johnsson, K. / Hovius, R. / Gorszka, K.I. / Pojer, F. | ||||||
 Citation Citation | Journal: Neuron / Year: 2015 Title: Reduction of Neuropathic and Inflammatory Pain through Inhibition of the Tetrahydrobiopterin Pathway. Authors: Latremoliere, A. / Latini, A. / Andrews, N. / Cronin, S.J. / Fujita, M. / Gorska, K. / Hovius, R. / Romero, C. / Chuaiphichai, S. / Painter, M. / Miracca, G. / Babaniyi, O. / Remor, A.P. / ...Authors: Latremoliere, A. / Latini, A. / Andrews, N. / Cronin, S.J. / Fujita, M. / Gorska, K. / Hovius, R. / Romero, C. / Chuaiphichai, S. / Painter, M. / Miracca, G. / Babaniyi, O. / Remor, A.P. / Duong, K. / Riva, P. / Barrett, L.B. / Ferreiros, N. / Naylor, A. / Penninger, J.M. / Tegeder, I. / Zhong, J. / Blagg, J. / Channon, K.M. / Johnsson, K. / Costigan, M. / Woolf, C.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xwy.cif.gz | 216.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xwy.ent.gz | 173.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4xwy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xw/4xwyftp://data.pdbj.org/pub/pdb/validation_reports/xw/4xwy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4hwkS S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / SPR Mass: 29891.414 Da / Num. of mol.: 4 / Fragment: residues 15-275 Source method: isolated from a genetically manipulated source Details: The N-termius" MHHHHHHENLYFQG" was added for purification M15 is original start codon of the protein residues 1-17 "MHHHHHHENLYFQGMEG" and 275 "K" are not resolved in the structure Source: (gene. exp.) Homo sapiens (human) / Gene: SPR / Plasmid: pET9a / Production host:  Escherichia coli bl21(de3) (bacteria) Escherichia coli bl21(de3) (bacteria)References: UniProt: P35270, sepiapterin reductase (L-erythro-7,8-dihydrobiopterin forming) #2: Chemical | ChemComp-NDP / Nicotinamide adenine dinucleotide phosphate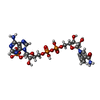  Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3#3: Chemical | ChemComp-43O /   Mass: 262.304 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H18N2O3 Mass: 262.304 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H18N2O3#4: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 249 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.58 Å3/Da / Density % sol: 73.12 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 2% W/V PEG1000, 2.5% V/V glycerol, 1.7 M Ammonium sulfate, 0.1 M Hepes PH range: 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 0.99999 Å / Beamline: X06SA / Wavelength: 0.99999 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 11, 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: SI(111) MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.99999 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.35→125.435 Å / Num. all: 89220 / Num. obs: 89220 / % possible obs: 100 % / Redundancy: 11.2 % / Biso Wilson estimate: 55.8 Å2 / Rpim(I) all: 0.03 / Rrim(I) all: 0.1 / Rsym value: 0.096 / Net I/av σ(I): 6.536 / Net I/σ(I): 15.8 / Num. measured all: 998773 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
-Phasing
| Phasing | Method: molecular replacement | ||||||
|---|---|---|---|---|---|---|---|
| Phasing MR | R rigid body: 0.555
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4HWK Resolution: 2.35→125.435 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.937 / WRfactor Rfree: 0.1983 / WRfactor Rwork: 0.1699 / FOM work R set: 0.8433 / SU B: 5.423 / SU ML: 0.126 / SU R Cruickshank DPI: 0.1789 / SU Rfree: 0.1629 / Cross valid method: FREE R-VALUE / σ(F): 0 / ESU R: 0.179 / ESU R Free: 0.163 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 200.07 Å2 / Biso mean: 48.374 Å2 / Biso min: 21.32 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.296 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.35→125.435 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.35→2.411 Å / Total num. of bins used: 20
|
