 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xhl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of S. cerevisiae Hrr25 1-394 (K38R mutant) | ||||||
 Components Components | Casein kinase I homolog HRR25 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / casein kinase /  monopolin / TRANSFERASE-TRANSFERASE INHIBITOR complex monopolin / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of vesicle fusion with Golgi apparatus / regulation of protein localization by the Cvt pathway / monopolin complex / positive regulation of clathrin-dependent endocytosis / spindle attachment to meiosis I kinetochore / regulation of ER to Golgi vesicle-mediated transport / meiotic chromosome segregation / tRNA wobble uridine modification / cellular bud tip / regulation of autophagosome assembly ...regulation of vesicle fusion with Golgi apparatus / regulation of protein localization by the Cvt pathway / monopolin complex / positive regulation of clathrin-dependent endocytosis / spindle attachment to meiosis I kinetochore / regulation of ER to Golgi vesicle-mediated transport / meiotic chromosome segregation / tRNA wobble uridine modification / cellular bud tip / regulation of autophagosome assembly / pexophagy / cellular bud neck / spindle pole body / preribosome, small subunit precursor / Major pathway of rRNA processing in the nucleolus and cytosol / chromosome, centromeric region / ribosomal large subunit biogenesis / ribosomal small subunit biogenesis / endocytosis / regulation of protein localization / protein tyrosine kinase activity / non-specific serine/threonine protein kinase / protein kinase activity / phosphorylation / protein serine kinase activity / DNA repair / protein serine/threonine kinase activity / nucleolus / Golgi apparatus / signal transduction / nucleoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.01 Å | ||||||
 Authors Authors | Ye, Q. / Corbett, K.D. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Embo J. / Year: 2016 Title: Structure of the Saccharomyces cerevisiae Hrr25:Mam1 monopolin subcomplex reveals a novel kinase regulator. Authors: Ye, Q. / Ur, S.N. / Su, T.Y. / Corbett, K.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xhl.cif.gz | 158.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xhl.ent.gz | 124.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4xhl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xh/4xhlftp://data.pdbj.org/pub/pdb/validation_reports/xh/4xhl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4xh0SC  4xhgC  4xhhC  5cyzC  5czoC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 45934.566 Da / Num. of mol.: 1 / Fragment: UNP residues 1-394 / Mutation: K38R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Gene: HRR25 / Production host:  Escherichia coli (E. coli) Escherichia coli (E. coli)References: UniProt: P29295, non-specific serine/threonine protein kinase | ||
|---|---|---|---|
| #2: Chemical | ChemComp-CKI / 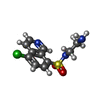  Mass: 285.750 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12ClN3O2S Mass: 285.750 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12ClN3O2S | ||
| #3: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.81 Å3/Da / Density % sol: 56.24 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 11 Details: 0.1 M CAPS pH 11, 0.2 M lithium sulfate, and 1.5-1.6 M ammonium sulfate PH range: 11 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-E / Wavelength: 0.9792 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Oct 15, 2012 |
| Radiation | Monochromator: double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 3.01→119.08 Å / Num. obs: 10857 / % possible obs: 99.5 % / Redundancy: 10.6 % / Rmerge(I) obs: 0.11 / Net I/σ(I): 16.8 |
| Reflection shell | Resolution: 3.01→3.17 Å / Redundancy: 10.7 % / Rmerge(I) obs: 1.252 / Mean I/σ(I) obs: 2 / % possible all: 99.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4XH0 Resolution: 3.01→119.08 Å / SU ML: 0.42 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 32.21 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.01→119.08 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
