Entry Database : PDB / ID : 4s2tTitle Crystal structure of X-prolyl aminopeptidase from Caenorhabditis elegans: a cytosolic enzyme with a di-nuclear active site Keywords / / / / Function / homology Biological species Caenorhabditis elegans (invertebrata)Method / / / Resolution : 2.15 Å Authors Iyer, S. / La-Borde, P. / Payne, K.A.P. / Parsons, M.R. / Turner, A.J. / Isaac, R.E. / Acharya, K.R. Journal : FEBS Open Bio / Year : 2015Title : Crystal structure of X-prolyl aminopeptidase from Caenorhabditis elegans: A cytosolic enzyme with a di-nuclear active site.Authors : Iyer, S. / La-Borde, P.J. / Payne, K.A. / Parsons, M.R. / Turner, A.J. / Isaac, R.E. / Acharya, K.R. History Deposition Jan 22, 2015 Deposition site / Processing site Revision 1.0 Apr 22, 2015 Provider / Type Revision 1.1 May 6, 2015 Group Revision 1.2 Nov 22, 2017 Group / Refinement description / Category / softwareRevision 2.0 Nov 15, 2023 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Database references / Derived calculations / Refinement description Category atom_site / chem_comp_atom ... atom_site / chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / pdbx_unobs_or_zero_occ_atoms / struct_conn / struct_conn_type / struct_ncs_dom_lim / struct_ref_seq_dif / struct_site Item _atom_site.auth_atom_id / _atom_site.label_atom_id ... _atom_site.auth_atom_id / _atom_site.label_atom_id / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_asym_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn_type.id / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.beg_label_asym_id / _struct_ncs_dom_lim.beg_label_comp_id / _struct_ncs_dom_lim.beg_label_seq_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ncs_dom_lim.end_label_asym_id / _struct_ncs_dom_lim.end_label_comp_id / _struct_ncs_dom_lim.end_label_seq_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 2.1 Apr 3, 2024 Group / Category
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords metalloprotease /
metalloprotease /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj Assembly
Assembly

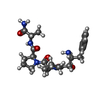
 Type: Peptide-like
Type: Peptide-like
 Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn
 Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 352 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 352 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I24 / Wavelength: 0.968 Å
/ Beamline: I24 / Wavelength: 0.968 Å Processing
Processing