 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4rdu: Crystal structure of a distal-less homeobox protein 5 (Dlx5) from... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4rdu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a distal-less homeobox protein 5 (Dlx5) from Homo sapiens at 1.85 A resolution | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / PF00046 family /  Structural Genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-BIOLOGY / TRANSCRIPTION / TRANSCRIPTION-DNA complex / Partnership for Stem Cell Biology / STEMCELL Structural Genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-BIOLOGY / TRANSCRIPTION / TRANSCRIPTION-DNA complex / Partnership for Stem Cell Biology / STEMCELL | ||||||
| Function / homology |  Function and homology information Function and homology informationinterneuron axon guidance / olfactory pit development / HMG box domain binding / anatomical structure formation involved in morphogenesis / olfactory bulb interneuron differentiation / endochondral ossification / embryonic limb morphogenesis / face morphogenesis / inner ear morphogenesis / roof of mouth development ...interneuron axon guidance / olfactory pit development / HMG box domain binding / anatomical structure formation involved in morphogenesis / olfactory bulb interneuron differentiation / endochondral ossification / embryonic limb morphogenesis / face morphogenesis / inner ear morphogenesis / roof of mouth development / BMP signaling pathway / epithelial cell differentiation / skeletal system development / positive regulation of epithelial cell proliferation / osteoblast differentiation / positive regulation of canonical Wnt signaling pathway / Regulation of RUNX2 expression and activity / sequence-specific double-stranded DNA binding / nervous system development / DNA-binding transcription activator activity, RNA polymerase II-specific / cell population proliferation / cell differentiation / transcription cis-regulatory region binding / DNA-binding transcription factor activity, RNA polymerase II-specific / RNA polymerase II cis-regulatory region sequence-specific DNA binding / chromatin / positive regulation of gene expression / regulation of transcription by RNA polymerase II / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.85 Å | ||||||
 Authors Authors | Joint Center for Structural Genomics (JCSG) / Partnership for Stem Cell Biology (STEMCELL) | ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of a distal-less homeobox protein 5 (Dlx5) from Homo sapiens at 1.85 A resolution Authors: Joint Center for Structural Genomics (JCSG) / Partnership for Stem Cell Biology (STEMCELL) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4rdu.cif.gz | 132 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4rdu.ent.gz | 99.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4rdu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rd/4rduftp://data.pdbj.org/pub/pdb/validation_reports/rd/4rdu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ig7S S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |








-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 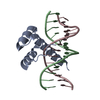
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Refine code: 4
NCS ensembles :
|
-Components
| #1: Protein | Mass: 7663.090 Da / Num. of mol.: 2 / Fragment: DNA binding residues 137-198 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BC006226, DLX5 / Plasmid: pET28bGB1Tev / Production host:  Escherichia Coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P56178 Escherichia Coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P56178#2: DNA chain | ( Mass: 4279.804 Da / Num. of mol.: 4 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2OSequence details | THE CONSTRUCT (135-198) WAS EXPRESSED WITH A PURIFICATION TAG MGSDKIHHHHHHENLYFQG. THE TAG WAS ...THE CONSTRUCT (135-198) WAS EXPRESSED WITH A PURIFICATI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.05 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 6 Details: 0.08M sodium chloride, 45.00% 2-methyl-2,4-pentanediol, 0.012M spermine tetrahydrochloride, 0.1M sodium cacodylate pH 6.0, NANODROP, VAPOR DIFFUSION, SITTING DROP, temperature 289K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL14-1 / Wavelength: 1 / Beamline: BL14-1 / Wavelength: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Jul 24, 2014 Details: Vertical focusing mirror; double crystal Si(111) monochromator | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: double crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.85→22.303 Å / Num. all: 23348 / Num. obs: 23348 / % possible obs: 95.5 % / Redundancy: 1.9 % / Biso Wilson estimate: 33.646 Å2 / Rsym value: 0.046 / Net I/σ(I): 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1IG7 Resolution: 1.85→22.303 Å / Cor.coef. Fo:Fc: 0.971 / Cor.coef. Fo:Fc free: 0.956 / Occupancy max: 1 / Occupancy min: 0.33 / SU B: 6.492 / SU ML: 0.102 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.153 / ESU R Free: 0.142 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 2. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS. 3.TLS GROUPS WERE ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 2. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS. 3.TLS GROUPS WERE ASSIGNED WITH THE AID OF THE TLSMD (MOTION DETERMINATION) SERVER. 4. WATERS WERE EXCLUDED FROM TLS ASSIGNMENT.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 158.2 Å2 / Biso mean: 47.0789 Å2 / Biso min: 20.44 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→22.303 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.85→1.897 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
