- PDB-4qne: Inosine 5'-monophosphate dehydrogenase from Vibrio cholerae, dele... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4qne
Title
Inosine 5'-monophosphate dehydrogenase from Vibrio cholerae, deletion mutant, in complex with NAD and IMP
Components
Inosine 5'-monophosphate dehydrogenase
Keywords
OXIDOREDUCTASE / structural genomics / IMPDH / NIAID / National Institute of Allergy and Infectious Diseases / Center for Structural Genomics of Infectious Diseases / CSGID / IMP / NAD acidic form
Function / homology
Function and homology information
IMP dehydrogenase activity / IMP dehydrogenase / GMP biosynthetic process / GTP biosynthetic process / nucleotide binding / metal ion binding / cytoplasm Similarity search - Function
IMP dehydrogenase / GMP reductase domain / Inosine-5'-monophosphate dehydrogenase / IMP dehydrogenase / GMP reductase, conserved site / IMP dehydrogenase / GMP reductase signature. / IMP dehydrogenase/GMP reductase / IMP dehydrogenase / GMP reductase domain / CBS domain superfamily / Domain in cystathionine beta-synthase and other proteins. / CBS domain / CBS domain ...IMP dehydrogenase / GMP reductase domain / Inosine-5'-monophosphate dehydrogenase / IMP dehydrogenase / GMP reductase, conserved site / IMP dehydrogenase / GMP reductase signature. / IMP dehydrogenase/GMP reductase / IMP dehydrogenase / GMP reductase domain / CBS domain superfamily / Domain in cystathionine beta-synthase and other proteins. / CBS domain / CBS domain / CBS domain profile. / Aldolase class I / Aldolase-type TIM barrel / TIM Barrel / Alpha-Beta Barrel / Alpha Beta Similarity search - Domain/homology
Resolution: 2.32→47.5 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.931 / SU B: 17.244 / SU ML: 0.206 / Cross valid method: THROUGHOUT / ESU R: 0.39 / ESU R Free: 0.275 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.27019
1553
5 %
RANDOM
Rwork
0.1963
-
-
-
obs
0.2
29325
99.04 %
-
all
-
30878
-
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords OXIDOREDUCTASE /
OXIDOREDUCTASE /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly

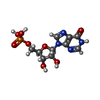
 Mass: 348.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N4O8P
Mass: 348.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N4O8P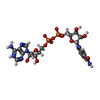
 Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2
Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2
 Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 19-BM / Wavelength: 0.9792 Å
/ Beamline: 19-BM / Wavelength: 0.9792 Å Processing
Processing