| Entry | Database: PDB / ID: 4l5l
|
|---|
| Title | Crystal structure of 26 kDa GST of Clonorchis sinensis in P212121 symmetry |
|---|
 Components Components | Putative glutathione transferase Glutathione S-transferase Glutathione S-transferase |
|---|
 Keywords Keywords | TRANSFERASE / GSH Binding |
|---|
| Function / homology |  Function and homology information Function and homology information
Glutathione S-transferase, C-terminal domain / Glutathione S-transferase, N-terminal domain / Glutathione transferase family / Glutathione S-transferase Yfyf (Class Pi); Chain A, domain 2 - #10 / Glutathione S-transferase, C-terminal / Glutathione S-transferase Yfyf (Class Pi); Chain A, domain 2 / Soluble glutathione S-transferase N-terminal domain profile. / Glutathione S-transferase, C-terminal-like / Soluble glutathione S-transferase C-terminal domain profile. / Glutathione S-transferase, N-terminal ...Glutathione S-transferase, C-terminal domain / Glutathione S-transferase, N-terminal domain / Glutathione transferase family / Glutathione S-transferase Yfyf (Class Pi); Chain A, domain 2 - #10 / Glutathione S-transferase, C-terminal / Glutathione S-transferase Yfyf (Class Pi); Chain A, domain 2 / Soluble glutathione S-transferase N-terminal domain profile. / Glutathione S-transferase, C-terminal-like / Soluble glutathione S-transferase C-terminal domain profile. / Glutathione S-transferase, N-terminal / Glutathione S-transferase, C-terminal domain superfamily / Glutaredoxin / Glutaredoxin / Thioredoxin-like superfamily / Up-down Bundle / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Clonorchis sinensis (oriental liver fluke) Clonorchis sinensis (oriental liver fluke) |
|---|
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.2 Å |
|---|
 Authors Authors | Chung, Y.J. / Han, Y.H. |
|---|
 Citation Citation | Journal: Biochem.Biophys.Res.Commun. / Year: 2013
Title: Crystal structures of 26kDa Clonorchis sinensis glutathione S-transferase reveal zinc binding and putative metal binding.
Authors: Han, Y.H. / Hong, S.J. / Cheong, H.K. / Chung, Y.J. |
|---|
| History | | Deposition | Jun 11, 2013 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Sep 4, 2013 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 15, 2017 | Group: Refinement description / Category: software
Item: _software.classification / _software.contact_author ..._software.classification / _software.contact_author / _software.contact_author_email / _software.date / _software.language / _software.location / _software.name / _software.type / _software.version |
|---|
| Revision 1.2 | Mar 20, 2024 | Group: Data collection / Database references / Derived calculations
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj

 Assembly
Assembly

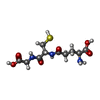
 Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
 Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM
Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 7A (6B, 6C1) / Wavelength: 1.23985 Å
/ Beamline: 7A (6B, 6C1) / Wavelength: 1.23985 Å Processing
Processing