 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ku5: Crystal Structures of C143S Xanthomonas campestris OleA with Boun... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ku5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structures of C143S Xanthomonas campestris OleA with Bound Lauric Acid and Lauroyl-CoA | ||||||
 Components Components | 3-oxoacyl-[ACP] synthase III | ||||||
 Keywords Keywords |  TRANSFERASE / Thiolase TRANSFERASE / Thiolase | ||||||
| Function / homology |  Function and homology information Function and homology informationacyl-CoA:acyl-CoA alkyltransferase / secondary metabolite biosynthetic process / 3-oxoacyl-[acyl-carrier-protein] synthase activity / fatty acid biosynthetic process / metal ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Xanthomonas campestris pv. campestris (bacteria) Xanthomonas campestris pv. campestris (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.17 Å | ||||||
 Authors Authors | Goblirsch, B.R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2016 Title: Substrate Trapping in Crystals of the Thiolase OleA Identifies Three Channels That Enable Long Chain Olefin Biosynthesis. Authors: Goblirsch, B.R. / Jensen, M.R. / Mohamed, F.A. / Wackett, L.P. / Wilmot, C.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ku5.cif.gz | 284 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ku5.ent.gz | 227.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4ku5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ku/4ku5ftp://data.pdbj.org/pub/pdb/validation_reports/ku/4ku5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ktiC  4ktmC  4ku2C  4ku3C  3rowS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 38826.379 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Xanthomonas campestris pv. campestris (bacteria)Strain: ATCC 33913 / NCPPB 528 / LMG 568 / Gene: fabH, XCC0212 / Production host: Escherichia coli (E. coli) / References: UniProt: Q8PDX2 |
|---|
-Non-polymers , 5 types, 383 molecules 

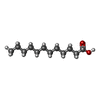


| #2: Chemical | ChemComp-PEG / Diethylene glycol Mass: 106.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C4H10O3#3: Chemical | ChemComp-PO4 / | Phosphate Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#4: Chemical | ChemComp-DAO / | Lauric acid Mass: 200.318 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H24O2 Mass: 200.318 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H24O2#5: Chemical | ChemComp-DCC / |  Mass: 949.837 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H58N7O17P3S Mass: 949.837 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H58N7O17P3S#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 375 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 4.2 Details: 18% PEG 8000, 80 mM potassium phosphate dibasic, 100 mM sodium citrate , pH 4.2, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-D / Wavelength: 1.03 Å / Beamline: 23-ID-D / Wavelength: 1.03 Å |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Nov 4, 2012 |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.03 Å / Relative weight: 1 |
| Reflection | Resolution: 2.17→50 Å / Num. all: 263108 / Num. obs: 263108 / % possible obs: 100 % / Observed criterion σ(F): -3 / Observed criterion σ(I): 0 / Redundancy: 6.8 % / Biso Wilson estimate: 29.7 Å2 / Rmerge(I) obs: 0.0109 / Net I/σ(I): 18.2 |
| Reflection shell | Resolution: 2.17→2.22 Å / Rmerge(I) obs: 0.575 / Mean I/σ(I) obs: 3.5 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 3ROW Resolution: 2.17→44.006 Å / SU ML: 0.2 / σ(F): 1.35 / Phase error: 20.67 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.22 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.17→44.006 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
