- PDB-4iyn: Structure of mitochondrial Hsp90 (TRAP1) with ADP-ALF4- -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4iyn
Title
Structure of mitochondrial Hsp90 (TRAP1) with ADP-ALF4-
Components
TNF receptor-associated protein 1
Keywords
CHAPERONE / ATP BINDING / ATPase / mitochondria
Function / homology
Function and homology information
nucleic acid metabolic process / ATP-dependent protein folding chaperone / unfolded protein binding / protein folding / mitochondrial inner membrane / calcium ion binding / protein kinase binding / magnesium ion binding / ATP hydrolysis activity / ATP binding / identical protein binding Similarity search - Function
Heat shock protein 90, C-terminal domain / Rossmann fold - #11260 / Ribosomal Protein S5; domain 2 - #80 / Ribosomal Protein S5; domain 2 / HSP90, C-terminal domain / Heat shock protein Hsp90, N-terminal / Heat shock protein Hsp90 family / Hsp90 protein / Histidine kinase-, DNA gyrase B-, and HSP90-like ATPase / Histidine kinase-like ATPase, C-terminal domain ...Heat shock protein 90, C-terminal domain / Rossmann fold - #11260 / Ribosomal Protein S5; domain 2 - #80 / Ribosomal Protein S5; domain 2 / HSP90, C-terminal domain / Heat shock protein Hsp90, N-terminal / Heat shock protein Hsp90 family / Hsp90 protein / Histidine kinase-, DNA gyrase B-, and HSP90-like ATPase / Histidine kinase-like ATPase, C-terminal domain / Heat Shock Protein 90 / Four Helix Bundle (Hemerythrin (Met), subunit A) / Histidine kinase-like ATPases / Histidine kinase/HSP90-like ATPase / Histidine kinase/HSP90-like ATPase superfamily / Ribosomal protein S5 domain 2-type fold / Up-down Bundle / Rossmann fold / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
ADENOSINE-5'-DIPHOSPHATE / TETRAFLUOROALUMINATE ION / : / Heat shock protein 75 kDa, mitochondrial Similarity search - Component
Biological species
Danio rerio (zebrafish)
Method
X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.312 Å
Resolution: 2.31→2.39 Å / Redundancy: 3.8 % / Mean I/σ(I) obs: 2.41 / Rsym value: 0.431 / % possible all: 91.1
-
Processing
Software
Name
Version
Classification
Blu-Ice
datacollection
SOLVE
phasing
PHENIX
(phenix.refine: dev_1214)
refinement
HKL-2000
datareduction
HKL-2000
datascaling
Refinement
Method to determine structure: SAD Starting model: SeMET derivative of TRAP1 with ADP-ALF4 Resolution: 2.312→29.811 Å / SU ML: 0.3 / σ(F): 1.34 / Phase error: 26.01 / Stereochemistry target values: ML
Rfactor
Num. reflection
% reflection
Rfree
0.2309
1994
2.99 %
Rwork
0.1912
-
-
obs
0.1924
66710
99.25 %
all
-
67214
-
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Refinement step
Cycle: LAST / Resolution: 2.312→29.811 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
9690
0
70
277
10037
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.012
10013
X-RAY DIFFRACTION
f_angle_d
1.148
13545
X-RAY DIFFRACTION
f_dihedral_angle_d
15.693
3805
X-RAY DIFFRACTION
f_chiral_restr
0.076
1500
X-RAY DIFFRACTION
f_plane_restr
0.005
1729
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.3115-2.3693
0.368
125
0.2806
4209
X-RAY DIFFRACTION
90
2.3693-2.4333
0.3102
135
0.245
4647
X-RAY DIFFRACTION
100
2.4333-2.5049
0.2868
142
0.2443
4620
X-RAY DIFFRACTION
100
2.5049-2.5857
0.3227
141
0.2341
4606
X-RAY DIFFRACTION
100
2.5857-2.6781
0.2493
140
0.2304
4677
X-RAY DIFFRACTION
100
2.6781-2.7852
0.2875
142
0.2311
4604
X-RAY DIFFRACTION
100
2.7852-2.9119
0.2422
142
0.2242
4652
X-RAY DIFFRACTION
100
2.9119-3.0653
0.2873
142
0.215
4640
X-RAY DIFFRACTION
100
3.0653-3.2571
0.2489
142
0.2139
4642
X-RAY DIFFRACTION
100
3.2571-3.5082
0.2379
150
0.2058
4665
X-RAY DIFFRACTION
100
3.5082-3.8606
0.249
145
0.1808
4653
X-RAY DIFFRACTION
100
3.8606-4.4177
0.2085
149
0.1625
4676
X-RAY DIFFRACTION
100
4.4177-5.5599
0.1916
153
0.1662
4657
X-RAY DIFFRACTION
100
5.5599-29.8139
0.191
146
0.1707
4768
X-RAY DIFFRACTION
100
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
3.2154
-0.1378
-0.0183
2.5048
0.5895
1.1399
0.0613
-0.4012
0.0407
0.197
-0.0705
-0.2485
0.0905
0.1401
-0.0048
0.3585
-0.0462
0.0131
0.2952
-0.0105
0.3628
5.004
17.8335
59.276
2
1.7378
-0.8498
0.8623
2.1957
-0.7606
5.6498
0.0102
0.1573
0.0436
-0.3209
-0.0516
0.1755
-0.0931
0.0294
0.0474
0.4414
-0.0501
-0.0064
0.2773
-0.0464
0.5195
-4.8415
30.7353
32.5387
3
2.557
-1.4223
1.155
3.8512
-1.7713
4.2248
-0.4282
-0.1769
0.9558
-0.5958
-0.0645
0.7635
-0.7039
-0.7686
0.4337
0.7959
0.121
-0.4215
0.6243
-0.1097
1.0785
-26.2375
39.9439
13.1035
4
3.5812
-2.3585
-1.6549
3.0138
0.4063
1.507
-0.1172
0.4246
0.0927
-0.1136
-0.2282
0.6891
-0.2607
-0.5457
0.3587
0.7731
-0.029
-0.366
0.718
0.0531
0.7888
-33.2239
20.2293
-10.5235
5
4.6357
0.6102
0.1943
1.7597
0.2829
0.7461
0.1102
-0.7143
0.4181
0.3792
-0.2654
0.656
0.0874
-0.2127
0.1187
0.4448
-0.094
0.196
0.5131
-0.1228
0.6704
-23.1581
18.5013
65.3921
6
1.1772
-0.0295
-0.0532
2.2613
0.9182
4.6472
0.1167
0.0288
0.2635
0.0241
-0.0082
0.2424
-0.1687
-0.0055
-0.0992
0.2614
0.0201
0.0724
0.2691
0.0502
0.5095
-25.3664
5.1253
36.0595
7
3.911
1.0158
0.2733
4.2138
0.9917
7.5022
-0.3862
0.0435
-0.4009
-0.2013
0.2718
0.3251
0.52
0.0705
0.0803
0.3566
-0.0443
0.0343
0.2933
0.0998
0.3458
-17.4175
-5.2444
9.144
8
3.3423
-1.1948
0.4712
2.3841
-0.5388
4.8293
-0.0399
0.4144
0.0901
-0.5009
-0.0182
0.2027
-0.0442
0.2215
0.0694
0.6315
-0.1592
-0.0928
0.4747
0.0725
0.2868
-11.0456
6.1605
-13.3956
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
ChainAandresid85:310
2
X-RAY DIFFRACTION
2
ChainAandresid311:472
3
X-RAY DIFFRACTION
3
ChainAandresid473:566
4
X-RAY DIFFRACTION
4
ChainAandresid567:718
5
X-RAY DIFFRACTION
5
ChainBandresid85:310
6
X-RAY DIFFRACTION
6
ChainBandresid311:472
7
X-RAY DIFFRACTION
7
ChainBandresid473:566
8
X-RAY DIFFRACTION
8
ChainBandresid567:717
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords CHAPERONE /
CHAPERONE /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj






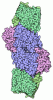


 Assembly
Assembly



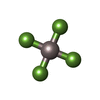


 Mass: 58.933 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Co
Mass: 58.933 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Co Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 102.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF4
Mass: 102.975 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF4 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Sample preparation
Sample preparation / Beamline: 8.3.1 / Wavelength: 1.115869 Å
/ Beamline: 8.3.1 / Wavelength: 1.115869 Å Processing
Processing