 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4gw9: Structure of a bacteriophytochrome and light-stimulated protomer ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4gw9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor | |||||||||
 Components Components | bacteriophytochrome | |||||||||
 Keywords Keywords |  SIGNALING PROTEIN / PHOTORECEPTOR / PAS/PAC SENSOR / BACTERIOPBHYTOCHROME / BACTERIOPHYTOCHROME PHOTOSENSORY AND C-TERMINAL OUTPUT TRANSDUCING DOMAINS / GENE REPRESSOR RPPPSR2 / photosensory core domain and PAS/PAC domain / Light signaling / PpsR2 SIGNALING PROTEIN / PHOTORECEPTOR / PAS/PAC SENSOR / BACTERIOPBHYTOCHROME / BACTERIOPHYTOCHROME PHOTOSENSORY AND C-TERMINAL OUTPUT TRANSDUCING DOMAINS / GENE REPRESSOR RPPPSR2 / photosensory core domain and PAS/PAC domain / Light signaling / PpsR2 | |||||||||
| Function / homology |  Function and homology information Function and homology informationdetection of visible light / photoreceptor activity / regulation of DNA-templated transcription / ATP bindingSimilarity search - Function | |||||||||
| Biological species |  Rhodopseudomonas palustris (phototrophic) Rhodopseudomonas palustris (phototrophic) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.9 Å | |||||||||
 Authors Authors | Bellini, D. / Papiz, M.Z. | |||||||||
 Citation Citation | Journal: Structure / Year: 2012 Title: Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Authors: Bellini, D. / Papiz, M.Z. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4gw9.cif.gz | 981 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4gw9.ent.gz | 847.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4gw9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gw/4gw9ftp://data.pdbj.org/pub/pdb/validation_reports/gw/4gw9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 72958.180 Da / Num. of mol.: 4 / Fragment: N-terminal 70 kDa fragment Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rhodopseudomonas palustris (phototrophic)Strain: CGA009 / Gene: RPA1537 / Plasmid: pET28a / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 (DE3) / References: UniProt: B3Q7C0, histidine kinase#2: Chemical | ChemComp-BLA / 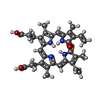  Mass: 582.646 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C33H34N4O6 Mass: 582.646 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C33H34N4O6#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 317 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 317 / Source method: isolated from a natural source / Formula: H2OSequence details | THE FULL LENGTH PROTEIN WITH N-HIS6TAG-(1-730) WAS EXPRESSED. HOWEVER, RESIDUES 636-730 WERE LOST ...THE FULL LENGTH PROTEIN WITH N-HIS6TAG-(1-730) WAS EXPRESSED. HOWEVER, RESIDUES 636-730 WERE LOST DURING PURIFICATI | Source details | THE PROTEIN SOURCE IS RHODOPSEUDOMONAS PALUSTRIS, STRAIN CGA009 AND THE GENE IS RPA1537. THIS GENE ...THE PROTEIN SOURCE IS RHODOPSEUD | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.69 Å3/Da / Density % sol: 66.7 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 8 Details: PROTEIN CONCENTRATION 20 MG/ML, 4% POLY-GAMMA-GLUTAMIC ACID POLYMER, 100 MM TRISHCL PH 8, 0.4 M NIACINAMIDE, 200 MM KBR, VAPOR DIFFUSION, SITTING DROP, temperature 292K |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 2M-F / Detector: PIXEL / Date: Feb 24, 2011 | ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 2.8→74 Å / Num. obs: 97789 / % possible obs: 97.7 % / Observed criterion σ(I): 0 / Redundancy: 3.4 % / Rsym value: 0.066 / Net I/σ(I): 9.6 | ||||||||||||||||||
| Reflection shell | Resolution: 2.8→2.87 Å / Redundancy: 3.1 % / Mean I/σ(I) obs: 1.4 / Rsym value: 0.63 / % possible all: 96.4 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.9→15 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.929 / SU B: 29.769 / SU ML: 0.269 / Cross valid method: THROUGHOUT / ESU R: 1.253 / ESU R Free: 0.352 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.01 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→2.972 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
