 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4fog: Crystal Structure of Mtb ThyA in Complex with 5-Fluoro-dUMP and 5... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4fog | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Mtb ThyA in Complex with 5-Fluoro-dUMP and 5-methyltetrahydrofolic acid | ||||||
 Components Components | Thymidylate synthase | ||||||
 Keywords Keywords | TRANSFERASE / Structural Genomics / TB Structural Genomics Consortium / TBSGC / thymidylate synthase | ||||||
| Function / homology |  Function and homology information Function and homology informationdUMP catabolic process / thymidylate synthase / thymidylate synthase activity / dTMP biosynthetic process / dTTP biosynthetic process / methylation / response to antibiotic / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.4 Å | ||||||
 Authors Authors | Reddy, M.C.M. / Bruning, J.B. / Harshbarger, W. / Sacchettini, J.C. / TB Structural Genomics Consortium (TBSGC) | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of binary and ternary complexes of thymidylate synthase (ThyA) from Mycobacterium tuberculosis: Insights into the selectivity and mode of inhibition Authors: Reddy, M.C.M. / Bruning, J.B. / Sacchettini, J.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4fog.cif.gz | 225.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4fog.ent.gz | 181.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4fog.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fo/4fogftp://data.pdbj.org/pub/pdb/validation_reports/fo/4fog | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4foaC  4foxC  3qj7S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / TS / TSase Mass: 29884.830 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Strain: H37Rv / Gene: MT2834, MTV002.29c, Rv2764c, thyA / Plasmid: pET28 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3)References: UniProt: P67044, UniProt: P9WFR9*PLUS, thymidylate synthase#2: Chemical | ChemComp-UFP / Fluorodeoxyuridylate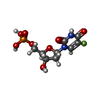  Type: DNA linking / Mass: 326.172 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H12FN2O8P / Comment: inhibitor*YM Type: DNA linking / Mass: 326.172 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H12FN2O8P / Comment: inhibitor*YM#3: Chemical | Levomefolic acid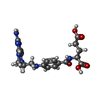  Mass: 459.456 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H25N7O6 Mass: 459.456 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H25N7O6#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 298 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 298 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.96 Å3/Da / Density % sol: 58.4 % |
|---|---|
| Crystal grow | Temperature: 298 K / pH: 6.4 Details: 2M ammonium sulfate, 0.1M imidazole pH 6.4, 5mM spermine, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 120 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.54 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Sep 12, 2009 / Details: OSMIC MIRRORS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: OSMIC MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 5.16 % / Number: 278963 / Rmerge(I) obs: 0.097 / Χ2: 1 / D res high: 2.4 Å / D res low: 48.65 Å / Num. obs: 53664 / % possible obs: 98 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell | ID: 1
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.4→48.65 Å / Num. obs: 53664 / % possible obs: 98 % / Observed criterion σ(I): 0 / Redundancy: 5.16 % / Biso Wilson estimate: 44.17 Å2 / Rmerge(I) obs: 0.097 / Rsym value: 0.097 / Net I/σ(I): 8.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 5.16 % / Rmerge(I) obs: 0.446 / Mean I/σ(I) obs: 2.9 / % possible all: 96.7 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3QJ7 Resolution: 2.4→42.94 Å / Occupancy max: 1 / Occupancy min: 0.34 / SU ML: 0.38 / Isotropic thermal model: isotropic / σ(F): 0 / Phase error: 26.9 / Stereochemistry target values: MLHL
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.95 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 64.66 Å2 / ksol: 0.38 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.32 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→42.94 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
