 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4emo | ||||||
|---|---|---|---|---|---|---|---|
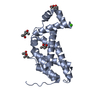
| Title | Crystal structure of the PH domain of SHARPIN | ||||||
 Components Components | Sharpin Lloyd Sharpin Lloyd Sharpin | ||||||
 Keywords Keywords | PROTEIN BINDING / Pleckstrin homology (PH) domain / LUBAC / SIPL1 / linear ubiquitin / HOIL-1L / HOIP | ||||||
| Function / homology |  Function and homology information Function and homology informationapoptotic nuclear changes / regulation of CD40 signaling pathway / protein linear polyubiquitination / LUBAC complex / regulation of tumor necrosis factor-mediated signaling pathway / Neurexins and neuroligins / TNFR1-induced proapoptotic signaling / keratinization / polyubiquitin modification-dependent protein binding / mitochondrion organization ...apoptotic nuclear changes / regulation of CD40 signaling pathway / protein linear polyubiquitination / LUBAC complex / regulation of tumor necrosis factor-mediated signaling pathway / Neurexins and neuroligins / TNFR1-induced proapoptotic signaling / keratinization / polyubiquitin modification-dependent protein binding / mitochondrion organization / TNFR1-induced NF-kappa-B signaling pathway / ubiquitin binding / Regulation of TNFR1 signaling / negative regulation of inflammatory response / ubiquitin-protein transferase activity / protein-macromolecule adaptor activity / proteasome-mediated ubiquitin-dependent protein catabolic process / positive regulation of canonical NF-kappaB signal transduction / defense response to bacterium / synapse / dendrite / protein-containing complex binding / identical protein binding / metal ion binding / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2 Å | ||||||
 Authors Authors | Stieglitz, B. / Haire, L.F. / Dikic, I. / Rittinger, K. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2012 Title: Structural Analysis of SHARPIN, a Subunit of a Large Multi-protein E3 Ubiquitin Ligase, Reveals a Novel Dimerization Function for the Pleckstrin Homology Superfold. Authors: Stieglitz, B. / Haire, L.F. / Dikic, I. / Rittinger, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4emo.cif.gz | 89.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4emo.ent.gz | 73.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4emo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/em/4emoftp://data.pdbj.org/pub/pdb/validation_reports/em/4emo | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Lloyd Sharpin / Shank-associated RH domain-interacting protein / Shank-interacting protein-like 1 / hSIPL1 Mass: 13355.524 Da / Num. of mol.: 4 / Mutation: L21M, L101M Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PSEC0216, SHARPIN, SIPL1 / Plasmid: pGex4T1 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q9H0F6 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q9H0F6#2: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.98 Å3/Da / Density % sol: 37.72 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.4 Details: 4M sodium formate, pH 7.4, vapor diffusion, sitting drop, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 77 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9799 Å / Beamline: I04 / Wavelength: 0.9799 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC / Detector: CCD / Date: May 29, 2009 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9799 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 3.9 % / Av σ(I) over netI: 17.17 / Number: 213897 / Rmerge(I) obs: 0.071 / Χ2: 0.95 / D res high: 2 Å / D res low: 30 Å / Num. obs: 55100 / % possible obs: 99.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→30 Å / Num. obs: 29888 / % possible obs: 99.1 % / Observed criterion σ(I): 2 / Redundancy: 3.9 % / Rmerge(I) obs: 0.071 / Χ2: 0.949 / Net I/σ(I): 10.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: SAD | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing dm | FOM : 0.73 / FOM acentric: 0.76 / FOM centric: 0.62 / Reflection: 17560 / Reflection acentric: 14320 / Reflection centric: 3240 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2→30 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.913 / WRfactor Rfree: 0.2697 / WRfactor Rwork: 0.2091 / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.8012 / SU B: 4.362 / SU ML: 0.125 / SU R Cruickshank DPI: 0.1973 / SU Rfree: 0.1864 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.186 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 94.58 Å2 / Biso mean: 34.5554 Å2 / Biso min: 14.85 Å2
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→30 Å
| |||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.048 Å / Total num. of bins used: 20
|
