 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4eei: Crystal Structure of Adenylosuccinate Lyase from Francisella tula... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4eei | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Adenylosuccinate Lyase from Francisella tularensis Complexed with AMP and Succinate | ||||||
 Components Components | Adenylosuccinate lyase | ||||||
 Keywords Keywords | LYASE / Structural Genomics / NIAID / National Institute of Allergy and Infectious Diseases / Center for Structural Genomics of Infectious Diseases / CSGID / alpha structure / AMP binding / beta-elimination / cytosole | ||||||
| Function / homology |  Function and homology informationadenylosuccinate lyase / N6-(1,2-dicarboxyethyl)AMP AMP-lyase (fumarate-forming) activity / (S)-2-(5-amino-1-(5-phospho-D-ribosyl)imidazole-4-carboxamido) succinate lyase (fumarate-forming) activity / 'de novo' AMP biosynthetic process / 'de novo' IMP biosynthetic process / nucleotide binding Function and homology informationadenylosuccinate lyase / N6-(1,2-dicarboxyethyl)AMP AMP-lyase (fumarate-forming) activity / (S)-2-(5-amino-1-(5-phospho-D-ribosyl)imidazole-4-carboxamido) succinate lyase (fumarate-forming) activity / 'de novo' AMP biosynthetic process / 'de novo' IMP biosynthetic process / nucleotide bindingSimilarity search - Function | ||||||
| Biological species |  Francisella tularensis subsp. tularensis SCHU S4 (bacteria) Francisella tularensis subsp. tularensis SCHU S4 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.921 Å | ||||||
 Authors Authors | Maltseva, N. / Kim, Y. / Shatsman, S. / Anderson, W.F. / Joachimiak, A. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of Adenylosuccinate Lyase from Francisella tularensis Complexed with AMP and Succinate Authors: Maltseva, N. / Kim, Y. / Shatsman, S. / Anderson, W.F. / Joachimiak, A. / Center for Structural Genomics of Infectious Diseases (CSGID) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4eei.cif.gz | 372.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4eei.ent.gz | 304.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4eei.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ee/4eeiftp://data.pdbj.org/pub/pdb/validation_reports/ee/4eei | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1c3uS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
| ||||||||||||
| Details | tetramer is generated by applying x,y,z, and y,x,-z+1 to asymmetric unit |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 50183.594 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Francisella tularensis subsp. tularensis SCHU S4 (bacteria)Strain: SCHU S4 / Schu 4 / Gene: FTT_0015, purB / Plasmid: pMCSG28 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3)-Magic / References: UniProt: Q5NIQ1, adenylosuccinate lyase |
|---|
-Non-polymers , 6 types, 626 molecules 


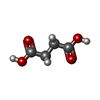


| #2: Chemical | Adenosine monophosphate Mass: 347.221 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM#3: Chemical | ChemComp-EDO / Ethylene glycol Mass: 62.068 Da / Num. of mol.: 17 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 17 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical |  Mass: 39.098 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: K#5: Chemical | ChemComp-SIN / | Succinic acid Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4#6: Chemical | Chloride Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 601 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.79 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 1.0M succinic acid pH7.0, 0.1M HEPES pH7.0, 1% PEG MME 2000, VAPOR DIFFUSION, SITTING DROP, temperature 289K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97915 Å / Beamline: 19-ID / Wavelength: 0.97915 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Mar 13, 2012 / Details: mirrors |
| Radiation | Monochromator: double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97915 Å / Relative weight: 1 |
| Reflection | Resolution: 1.92→50 Å / Num. all: 78595 / Num. obs: 78595 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 15.9 % / Biso Wilson estimate: 22.74 Å2 / Rmerge(I) obs: 0.105 / Net I/σ(I): 7.5 |
| Reflection shell | Resolution: 1.92→1.95 Å / Redundancy: 15.9 % / Rmerge(I) obs: 0.798 / Mean I/σ(I) obs: 4 / Num. unique all: 3857 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ID 1C3U Resolution: 1.921→39.331 Å / SU ML: 0.12 / Isotropic thermal model: mixed / Cross valid method: THROUGHOUT / σ(F): 0 / Phase error: 19.26 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.98 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 41.97 Å2 / ksol: 0.338 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.3 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.921→39.331 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
