 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4bxt: Crystal structure of the human metapneumovirus phosphoprotein tet... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4bxt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human metapneumovirus phosphoprotein tetramerization domain | ||||||
 Components Components | PHOSPHOPROTEIN P | ||||||
 Keywords Keywords |  VIRAL PROTEIN / TETRAMERIC PARALLEL COILED COIL VIRAL PROTEIN / TETRAMERIC PARALLEL COILED COIL | ||||||
| Function / homology | Phosphoprotein, pneumoviral / Pneumovirus phosphoprotein / RNA-dependent RNA polymerase activity / Phosphoprotein Function and homology information Function and homology information | ||||||
| Biological species |  HUMAN METAPNEUMOVIRUS HUMAN METAPNEUMOVIRUS | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.13 Å | ||||||
 Authors Authors | Leyrat, C. / Renner, M. / Harlos, K. / Grimes, J.M. | ||||||
 Citation Citation | Journal: Plos One / Year: 2013 Title: Solution and Crystallographic Structures of the Central Region of the Phosphoprotein from Human Metapneumovirus Authors: Leyrat, C. / Renner, M. / Harlos, K. / Grimes, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4bxt.cif.gz | 52.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4bxt.ent.gz | 38.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4bxt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bx/4bxtftp://data.pdbj.org/pub/pdb/validation_reports/bx/4bxt | HTTPS FTP |
|---|
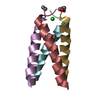
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 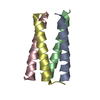
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.941234, 0.205036, -0.2684), Vector : |
-Components
| #1: Protein | Mass: 8773.052 Da / Num. of mol.: 8 / Fragment: TETRAMERIZATION DOMAIN, RESIDUES 158-237 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HUMAN METAPNEUMOVIRUS / Strain: SEROTYPE A1 (NL/1/00) / Plasmid: POPINF / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): ROSETTA2 / References: UniProt: Q91KZ5 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): ROSETTA2 / References: UniProt: Q91KZ5 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 39.7 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 25 % PEG 3350, 100 MM HEPES PH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.92001 / Beamline: I04 / Wavelength: 0.92001 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: May 22, 2013 |
| Radiation | Monochromator: DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92001 Å / Relative weight: 1 |
| Reflection | Resolution: 3.13→66.91 Å / Num. obs: 3602 / % possible obs: 90.3 % / Observed criterion σ(I): 1.5 / Redundancy: 10.5 % / Biso Wilson estimate: 77.44 Å2 / Rmerge(I) obs: 0.35 / Net I/σ(I): 6.4 |
| Reflection shell | Resolution: 3.13→3.21 Å / Redundancy: 6.1 % / Rmerge(I) obs: 1.35 / Mean I/σ(I) obs: 1.4 / % possible all: 57.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: SAXS-FILTERED ROSETTA FOLD-AND-DOCK AB INITIO MODEL Resolution: 3.13→46.42 Å / Cor.coef. Fo:Fc: 0.8587 / Cor.coef. Fo:Fc free: 0.7759 / Cross valid method: THROUGHOUT / σ(F): 0 / SU Rfree Blow DPI: 0.542
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.43 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.706 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.13→46.42 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.13→3.5 Å / Total num. of bins used: 5
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | S11: 0 Å ° / S12: 0 Å ° / S13: 0 Å ° / S21: 0 Å ° / S22: 0 Å ° / S23: 0 Å ° / S31: 0 Å ° / S32: 0 Å ° / S33: 0 Å ° / T11: 0 Å2 / T12: 0 Å2 / T13: 0 Å2 / T22: 0 Å2 / T23: 0 Å2 / T33: 0 Å2 / Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
