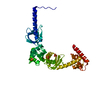
Entry Database : PDB / ID : 3zpgTitle Acinetobacter baumannii RibD, form 2 RIBD Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species ACINETOBACTER BAUMANNII (bacteria)Method / / Resolution : 1.99 Å Authors Dawson, A. / Trumper, P. / Chrysostomou, G. / Hunter, W.N. Journal : Acta Crystallogr.,Sect.F / Year : 2013Title : Structure of Diaminohydroxyphosphoribosylaminopyrimidine Deaminase/5-Amino-6-(5-Phosphoribosylamino)Uracil Reductase from Acinetobacter Baumannii.Authors : Dawson, A. / Trumper, P. / Chrysostomou, G. / Hunter, W.N. History Deposition Feb 27, 2013 Deposition site / Processing site Revision 1.0 Mar 27, 2013 Provider / Type Revision 1.1 Jun 12, 2013 Group Revision 1.2 Jun 28, 2017 Group / Category / Item Revision 1.3 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords HYDROLASE /
HYDROLASE /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj






 Assembly
Assembly






 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
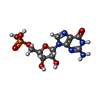
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 363.221 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N5O8P
Mass: 363.221 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H14N5O8P Mass: 88.019 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2O4
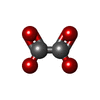
Mass: 88.019 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2O4 Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2
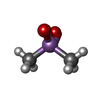
Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Sample preparation
Sample preparation Processing
Processing