 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3uyj: Crystal structure of JMJD5 catalytic core domain in complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3uyj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of JMJD5 catalytic core domain in complex with nickle and alpha-KG | ||||||
 Components Components | Lysine-specific demethylase 8 | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / jellyroll-like all beta fold / Demethylase / Nuclear OXIDOREDUCTASE / jellyroll-like all beta fold / Demethylase / Nuclear | ||||||
| Function / homology |  Function and homology information Function and homology information[protein]-arginine 3-hydroxylase / peptidyl-arginine 3-dioxygenase activity / histone H3K36 demethylase activity / Hydrolases; Acting on peptide bonds (peptidases) / Protein hydroxylation / aminopeptidase activity / methylated histone binding / regulation of signal transduction by p53 class mediator / circadian regulation of gene expression / protein destabilization ...[protein]-arginine 3-hydroxylase / peptidyl-arginine 3-dioxygenase activity / histone H3K36 demethylase activity / Hydrolases; Acting on peptide bonds (peptidases) / Protein hydroxylation / aminopeptidase activity / methylated histone binding / regulation of signal transduction by p53 class mediator / circadian regulation of gene expression / protein destabilization / G2/M transition of mitotic cell cycle / p53 binding / chromosome / fibroblast proliferation / endopeptidase activity / in utero embryonic development / negative regulation of DNA-templated transcription / chromatin binding / positive regulation of DNA-templated transcription / proteolysis / nucleoplasm / metal ion binding / nucleus / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.351 Å | ||||||
 Authors Authors | Su, X. / Li, H. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of JMJD5 catalytic core domain in complex with nickle and alpha-KG Authors: Su, X. / Li, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3uyj.cif.gz | 108.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3uyj.ent.gz | 88.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3uyj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uy/3uyjftp://data.pdbj.org/pub/pdb/validation_reports/uy/3uyj | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28683.609 Da / Num. of mol.: 2 / Fragment: Catalytic core domain, UNP residues 173-416 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: JMJD5, KDM8 / Plasmid: pET28b / Production host:  Escherichia coli (E. coli) / Strain (production host): Rossetta2(DE3) Escherichia coli (E. coli) / Strain (production host): Rossetta2(DE3)References: UniProt: Q8N371, [histone H3]-dimethyl-L-lysine36 demethylase #2: Chemical | Nickel  Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni#3: Chemical | Α-Ketoglutaric acid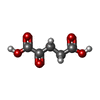  Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.81 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: evaporation / pH: 7.5 Details: 8% PEG3350, 0.1M HepesNa pH 7.5, EVAPORATION, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.97922 Å / Beamline: BL17U / Wavelength: 0.97922 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 8, 2011 / Details: mirrors |
| Radiation | Monochromator: Mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97922 Å / Relative weight: 1 |
| Reflection | Resolution: 2.35→20 Å / Num. all: 31174 / Num. obs: 30809 / % possible obs: 98.83 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 4.7 % / Biso Wilson estimate: 42.86 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 22.6 |
| Reflection shell | Resolution: 2.35→2.39 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.335 / % possible all: 99 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.351→19.994 Å / Occupancy max: 1 / Occupancy min: 1 / SU ML: 0.27 / σ(F): 0 / Phase error: 25.76 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 60.602 Å2 / ksol: 0.385 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.351→19.994 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
