 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3ubi: The Absence of Tertiary Interactions in a Self-Assembled DNA Crys... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3ubi | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Absence of Tertiary Interactions in a Self-Assembled DNA Crystal Structure | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  DNA / NANOTECHNOLOGY / DNA CROSSOVER / DESIGNED CRYSTAL LATTICE DNA / NANOTECHNOLOGY / DNA CROSSOVER / DESIGNED CRYSTAL LATTICE | ||||||
| Function / homology | DNA / DNA (> 10) Function and homology information Function and homology information | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 6.8046 Å | ||||||
 Authors Authors | Nguyen, N. / Birktoft, J.J. / Sha, R. / Wang, T. / Zheng, J. / Constantinou, P.E. / Ginell, S.L. / Chen, Y. / Mao, C. / Seeman, N.C. | ||||||
 Citation Citation | Journal: J.Mol.Recognit. / Year: 2012 Title: The absence of tertiary interactions in a self-assembled DNA crystal structure. Authors: Nguyen, N. / Birktoft, J.J. / Sha, R. / Wang, T. / Zheng, J. / Constantinou, P.E. / Ginell, S.L. / Chen, Y. / Mao, C. / Seeman, N.C. #1: Journal: Nature / Year: 2009Title: From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Authors: Zheng, J. / Birktoft, J.J. / Chen, Y. / Wang, T. / Sha, R. / Constantinou, P.E. / Ginell, S.L. / Mao, C. / Seeman, N.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3ubi.cif.gz | 36.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3ubi.ent.gz | 27 KB | Display | PDB format |
| PDBx/mmJSON format | 3ubi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ub/3ubiftp://data.pdbj.org/pub/pdb/validation_reports/ub/3ubi | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|






-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 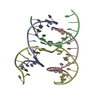
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 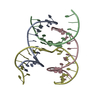
| ||||||||
| Unit cell |
| ||||||||
| Details | AUTHORS STATE THAT THE CRYSTAL IS AN INFINITE NETWORK MADE FROM THREE DNA STRANDS THAT SELF-ASSOCIATE. IN THE CURRENT ENTRY THE ASYMMETRIC UNIT IS COMPRISED OF 4 CHAINS, 3 OF WHICH ARE FRAGMENTS OF LONGER DNA STRANDS. APPLYING THE SPACE GROUP H3 SYMMETRY OPERATORS (X, Y, Z,), (-Y, X-Y, Z), AND (-X+Y,-X,Z) TO THE CONTENTS OF THE ASYMMETRIC UNIT GENERATES ONE TRIMERIC UNIT OF THE SELF-ASSOCIATED DNA NETWORK. ADDITIONAL DETAILS ABOUT THE CHEMICAL COMPOSITION AND ASSOCIATION ARE INCLUDED IN REMARK 400. |
-Components
| #1: DNA chain | Mass: 4884.168 Da / Num. of mol.: 1 Fragment: THE 16 RESIDUES IS FROM OF THE FIRST PART OF A DNA MOLECULE USED IN EXPERIMENT, SEE REMARK 400 FOR DETAILS. Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 5230.415 Da / Num. of mol.: 1 Fragment: SYMMETRICALLY- AND SEQUENTIALLY REPEATING UNIT OF A CIRCULAR DNA MOLECULES, SEE REMARK 400 FOR DETAILS. Source method: obtained synthetically |
| #3: DNA chain | Mass: 4311.801 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #4: DNA chain | Mass: 4550.957 Da / Num. of mol.: 1 Fragment: THE 15 RESIDUES IS FROM OF THE LAST PART OF A DNA MOLECULE USED IN EXPERIMENT, SEE REMARK 400 FOR DETAILS. Source method: obtained synthetically |
| Compound details | THE STRUCTURE UNIT IS GENERATED FROM 7 DNA STRANDS WHICH FORM A NETWORK UNIT WITH INTERNAL 3-FOLD ...THE STRUCTURE UNIT IS GENERATED FROM 7 DNA STRANDS WHICH FORM A NETWORK UNIT WITH INTERNAL 3-FOLD SYMMETRY. EXTENDING OF THE STRUCTURE UNIT IN 3-D SPACE RESULTS THE CRYSTAL AT R3 SPACE GROUP (REPRESENTE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: CRYSTALLIZATION CONDITIONS: GROWN BY VAPOR DIFFUSION WHILE TREATED WITH A CONTROLLED TEMPERATURE GRADIENT FROM 333 DEGS TO 293 DEGS., PH 8.5, VAPOR DIFFUSION, SITTING DROP |
|---|
-Data collection
| Diffraction | Mean temperature: 190 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 Å / Beamline: X25 / Wavelength: 1.1 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Details: GRAPHITE |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 6.805→49.069 Å / Num. all: 1413 / Num. obs: 1413 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 6.8046→49.069 Å / SU ML: -0 / Isotropic thermal model: Isotropic / σ(F): 0.24 / Phase error: 34.47 / Stereochemistry target values: MLHL
| ||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.47 Å / VDW probe radii: 0.8 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 0 Å2 / ksol: 0.126 e/Å3 | ||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 6.8046→49.069 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| LS refinement shell | Highest resolution: 6.8046 Å
|
