Entry Database : PDB / ID : 3hs8Title Intersectin 1-peptide-AP2 alpha ear complex Adaptor protein complex AP-2, alpha 2 subunit peptide from Intersectin-1, residues 840-851 Keywords / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Mus musculus (house mouse)Method / / / / Resolution : 1.9 Å Authors Vahedi-Faridi, A. / Pechstein, A. / Schaefer, J.G. / Saenger, W. / Haucke, V. Journal : Proc.Natl.Acad.Sci.USA / Year : 2010Title : Regulation of synaptic vesicle recycling by complex formation between intersectin 1 and the clathrin adaptor complex AP2.Authors : Pechstein, A. / Bacetic, J. / Vahedi-Faridi, A. / Gromova, K. / Sundborger, A. / Tomlin, N. / Krainer, G. / Vorontsova, O. / Schafer, J.G. / Owe, S.G. / Cousin, M.A. / Saenger, W. / Shupliakov, O. / Haucke, V. History Deposition Jun 10, 2009 Deposition site / Processing site Revision 1.0 Feb 23, 2010 Provider / Type Revision 1.1 Jul 13, 2011 Group / Refinement description / Version format complianceRevision 1.2 Nov 1, 2017 Group / Category Revision 1.3 Mar 20, 2024 Group / Database referencesCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / struct_ref_seq_dif Item / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords ENDOCYTOSIS / adaptor complex AP-2 /
ENDOCYTOSIS / adaptor complex AP-2 /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads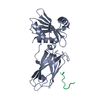










 PDBj
PDBj
















 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 14.2 / Wavelength: 0.91841 Å
/ Beamline: 14.2 / Wavelength: 0.91841 Å Processing
Processing