 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3evx: Crystal structure of the human E2-like ubiquitin-fold modifier co... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3evx | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human E2-like ubiquitin-fold modifier conjugating enzyme 1 (Ufc1). Northeast Structural Genomics Consortium target HR41 | |||||||||
 Components Components | Ufm1-conjugating enzyme 1 | |||||||||
 Keywords Keywords |  LIGASE / alpha-beta protein / Structural Genomics / PSI-2 / Protein Structure Initiative / Northeast Structural Genomics Consortium / NESG / Polymorphism / Ubl conjugation pathway LIGASE / alpha-beta protein / Structural Genomics / PSI-2 / Protein Structure Initiative / Northeast Structural Genomics Consortium / NESG / Polymorphism / Ubl conjugation pathway | |||||||||
| Function / homology |  Function and homology information Function and homology informationUFM1 conjugating enzyme activity / UFM1 transferase activity / protein ufmylation / protein K69-linked ufmylation / reticulophagy / response to endoplasmic reticulum stress / brain development / extracellular exosomeSimilarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.54 Å | |||||||||
 Authors Authors | Forouhar, F. / Abashidze, M. / Seetharaman, J. / Ho, C.K. / Janjua, H. / Cunningham, K. / Ma, L.-C. / Xiao, R. / Baran, M.C. / Acton, T.B. ...Forouhar, F. / Abashidze, M. / Seetharaman, J. / Ho, C.K. / Janjua, H. / Cunningham, K. / Ma, L.-C. / Xiao, R. / Baran, M.C. / Acton, T.B. / Rost, B. / Montelione, G.T. / Tong, L. / Hunt, J.F. / Northeast Structural Genomics Consortium (NESG) | |||||||||
 Citation Citation | Journal: J.STRUCT.FUNCT.GENOM. / Year: 2009 Title: NMR and X-RAY structures of human E2-like ubiquitin-fold modifier conjugating enzyme 1 (UFC1) reveal structural and functional conservation in the metazoan UFM1-UBA5-UFC1 ubiquination pathway. Authors: Liu, G. / Forouhar, F. / Eletsky, A. / Atreya, H.S. / Aramini, J.M. / Xiao, R. / Huang, Y.J. / Abashidze, M. / Seetharaman, J. / Liu, J. / Rost, B. / Acton, T. / Montelione, G.T. / Hunt, J.F. / Szyperski, T. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3evx.cif.gz | 139.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3evx.ent.gz | 113.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3evx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ev/3evxftp://data.pdbj.org/pub/pdb/validation_reports/ev/3evx | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 20698.234 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CGI-126, HSPC155, UFC1 / Plasmid: pET21 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3)+Magic / References: UniProt: Q9Y3C8 Escherichia coli (E. coli) / Strain (production host): BL21(DE3)+Magic / References: UniProt: Q9Y3C8#2: Chemical | ChemComp-SCN / Thiocyanate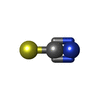  Mass: 58.082 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CNS#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 169 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 169 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.5 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 5 Details: Protein solution: 10 mM Tris (pH 7.5), 100 mM sodium chloride, and 5 mM DTT. Reservoir solution:100mM Sodium acetate, 18% PEG8000, 100mM (NH4)SCN, 50mM LiSCN, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X4A / Wavelength: 0.97898 Å / Beamline: X4A / Wavelength: 0.97898 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Aug 10, 2007 / Details: mirrors |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97898 Å / Relative weight: 1 |
| Reflection | Resolution: 2.54→30 Å / Num. all: 23056 / Num. obs: 22065 / % possible obs: 95.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.7 % / Biso Wilson estimate: 23.5 Å2 / Rmerge(I) obs: 0.081 / Rsym value: 0.066 / Net I/σ(I): 14.11 |
| Reflection shell | Resolution: 2.54→2.64 Å / Redundancy: 3.5 % / Rmerge(I) obs: 0.198 / Mean I/σ(I) obs: 5.8 / Num. unique all: 2496 / Rsym value: 0.149 / % possible all: 97.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.54→19.96 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 77510.06 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / σ(I): 2 / Stereochemistry target values: Engh & Huber Details: This crystal structure was initially refined in spacegroup P2(1), which gave canonical merging statistics during data processing that were very similar to those obtained in the lower ...Details: This crystal structure was initially refined in spacegroup P2(1), which gave canonical merging statistics during data processing that were very similar to those obtained in the lower symmetry spacegroup P1. However, the refinement of the structure was better in the latter spacegroup based on a 1.9% reduction in free R-factor at 2.54 for the same starting model when applying strong non-crystallographic symmetry (NCS) restraints (and with the reflections in the free set chosen in thin shells to avoid possible symmetry bias). The value of the free R-factor increased if NCS restraints were weakened, suggesting at most minimal conformational differences between protomers. However, the refined orientation of the NCS operator is inclined 0.04 degrees from the unit cell axis. This deviation from exact P2(1) symmetry is likely to account for the improved refinement statistics obtained in spacegroup P1, while its small size is consistent with the good merging statistics observed in spacegroup P2(1).
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 48.2457 Å2 / ksol: 0.45 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.9 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.54→19.96 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.54→2.63 Å / Rfactor Rfree error: 0.03 / Total num. of bins used: 10
|
