 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3bkd: High resolution Crystal structure of Transmembrane domain of M2 p... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bkd | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution Crystal structure of Transmembrane domain of M2 protein | ||||||
 Components Components | Transmembrane Domain of Matrix protein M2 | ||||||
 Keywords Keywords |  VIRAL PROTEIN / MEMBRANE PROTEIN / Proton channel / M2TM / Influenza A virus M2 Protein VIRAL PROTEIN / MEMBRANE PROTEIN / Proton channel / M2TM / Influenza A virus M2 Protein | ||||||
| Function / homology |  Function and homology information Function and homology informationsuppression by virus of host autophagy / : / proton transmembrane transporter activity / : / protein complex oligomerization / monoatomic ion channel activity / membrane => GO:0016020 / host cell plasma membrane / virion membrane Similarity search - Function | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Stouffer, A.L. / Acharya, R. / Salom, D. | ||||||
 Citation Citation | Journal: Nature / Year: 2008 Title: Structural basis for the function and inhibition of an influenza virus proton channel Authors: Stouffer, A.L. / Acharya, R. / Salom, D. / Levine, A.S. / Di Costanzo, L. / Soto, C.S. / Tereshko, V. / Nanda, V. / Stayrook, S. / DeGrado, W.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bkd.cif.gz | 54.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bkd.ent.gz | 42.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3bkd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bk/3bkdftp://data.pdbj.org/pub/pdb/validation_reports/bk/3bkd | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Beg auth comp-ID: SER / Beg label comp-ID: SER / End auth comp-ID: NH2 / End label comp-ID: NH2 / Refine code: 6 / Auth seq-ID: 22 - 47 / Label seq-ID: 1 - 26
NCS ensembles :
|
-Components
| #1: Protein/peptide | Mass: 2793.236 Da / Num. of mol.: 8 / Fragment: residues 22-46 / Source method: obtained synthetically Details: Chemically synthesized; Transmembrane domain of M2 protein from Influenza A virus References: UniProt: Q9Q0P0 #2: Sugar | ChemComp-BOG / Octyl glucoside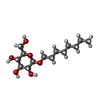  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 6 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 6Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM #3: Chemical | Chloride  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-PEG / Diethylene glycol  Mass: 106.120 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H10O3#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 53.91 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.3 Details: Protein solution: 0.8mM protein, 32mM n-octyl-beta-D-glucopyranoside and 5%v/v xylitol. Reservoir solution: 50mM Tris-Hcl, 500mM MgCl2, 21% PEG 350 MME, pH 7.3, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 0.97853 Å / Beamline: X25 / Wavelength: 0.97853 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Oct 1, 2001 |
| Radiation | Monochromator: Si 111 Channel / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97853 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. all: 15355 / Num. obs: 14567 / % possible obs: 94.9 % / Observed criterion σ(I): 1 / Redundancy: 3.4 % / Biso Wilson estimate: 37.1 Å2 / Rmerge(I) obs: 0.062 / Net I/σ(I): 22.4 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 2 % / Rmerge(I) obs: 0.305 / Mean I/σ(I) obs: 1.9 / Num. unique all: 777 / % possible all: 75.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Amantandine-bound M2TM; G34A mutant Resolution: 2.05→20 Å / Cor.coef. Fo:Fc: 0.937 / Cor.coef. Fo:Fc free: 0.915 / SU B: 9.819 / SU ML: 0.135 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.23 / ESU R Free: 0.199 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.117 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.05→2.103 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
