 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3b69 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | T cruzi Trans-sialidase complex with benzoylated NANA derivative | |||||||||
 Components Components | Trans-sialidase | |||||||||
 Keywords Keywords |  HYDROLASE / beta-propeller HYDROLASE / beta-propeller | |||||||||
| Function / homology |  Function and homology informationexo-alpha-sialidase activity / ganglioside catabolic process / oligosaccharide catabolic process / intracellular membrane-bounded organelle / membrane / cytoplasm Function and homology informationexo-alpha-sialidase activity / ganglioside catabolic process / oligosaccharide catabolic process / intracellular membrane-bounded organelle / membrane / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Trypanosoma cruzi (eukaryote) Trypanosoma cruzi (eukaryote) | |||||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.67 Å | |||||||||
 Authors Authors | Buschiazzo, A. | |||||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2008 Title: A new generation of specific Trypanosoma cruzi trans-sialidase inhibitors. Authors: Buchini, S. / Buschiazzo, A. / Withers, S.G. | |||||||||
| History |
| |||||||||
| Remark 999 | SEQUENCE AUTHORS INDICATE THAT THE SEQUENCE IN THE DATABASE IS INCORRECT |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3b69.cif.gz | 148.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3b69.ent.gz | 111 KB | Display | PDB format |
| PDBx/mmJSON format | 3b69.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b6/3b69ftp://data.pdbj.org/pub/pdb/validation_reports/b6/3b69 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ah2S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 71389.258 Da / Num. of mol.: 1 / Mutation: N59F, S496K, V497G, E521K, D594G, I598D, H600R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Trypanosoma cruzi (eukaryote) / Plasmid: pTrcHisA / Production host:  Escherichia coli (E. coli) / Strain (production host): TOP10 Escherichia coli (E. coli) / Strain (production host): TOP10References: UniProt: Q26966, UniProt: Q26964*PLUS, exo-alpha-sialidase |
|---|---|
| #2: Chemical | ChemComp-CL / Chloride  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #3: Sugar | ChemComp-BFN / 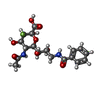  Type: D-saccharide, beta linking / Mass: 430.382 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23FN2O9 Type: D-saccharide, beta linking / Mass: 430.382 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H23FN2O9 |
| #4: Chemical | ChemComp-EPE / HEPES  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 378 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 378 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 51.89 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 10% PEG4000, 5% isopropanol, 0.1M HEPES, pH 7.5, vapor diffusion, hanging drop, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 110 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: May 8, 2007 / Details: VarimaxHF mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: confocal multilayer mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection twin | Type: hemihedral / Operator: l,-k,h / Fraction: 0.33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.67→64.599 Å / Num. all: 76475 / Num. obs: 76475 / % possible obs: 91.8 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 2.1 % / Rmerge(I) obs: 0.031 / Rsym value: 0.031 / Net I/σ(I): 16.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 0.356 / Cor.coef. Fo:Fc: 0.631 / Cor.coef. Io to Ic: 0.601
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2AH2 Resolution: 1.67→50 Å / Isotropic thermal model: restrained isotropic / Cross valid method: THROUGHOUT / σ(F): 3334 / Stereochemistry target values: Engh & Huber Details: Refinement was performed with the least squares target for hemihedrally twinned structures, as implemented in CNS v1.2 (twin operator l,-k,h; twin fraction 0.33)
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 52.03 Å2 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.606 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.67→50 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
