 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2y7w | ||||||
|---|---|---|---|---|---|---|---|
| Title | DntR Inducer Binding Domain | ||||||
 Components Components | LYSR-TYPE REGULATORY PROTEIN | ||||||
 Keywords Keywords |  TRANSCRIPTION TRANSCRIPTION | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  BURKHOLDERIA SP. (bacteria) BURKHOLDERIA SP. (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.89 Å | ||||||
 Authors Authors | Devesse, L. / Smirnova, I. / Lonneborg, R. / Kapp, U. / Brzezinski, P. / Leonard, G.A. / Dian, C. | ||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2011 Title: Crystal Structures of Dntr Inducer Binding Domains in Complex with Salicylate Offer Insights Into the Activation of Lysr-Type Transcriptional Regulators. Authors: Devesse, L. / Smirnova, I. / Lonneborg, R. / Kapp, U. / Brzezinski, P. / Leonard, G.A. / Dian, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2y7w.cif.gz | 175.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2y7w.ent.gz | 137.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2y7w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y7/2y7wftp://data.pdbj.org/pub/pdb/validation_reports/y7/2y7w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2y7kC  2y7pC  2y7rC  2y84C  1utbS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper:
|
-Components
| #1: Protein | Mass: 25974.059 Da / Num. of mol.: 4 / Fragment: INDUCER BINDING DOMAIN, RESIDUES 80-301 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) BURKHOLDERIA SP. (bacteria) / Strain: DNT / Production host: ESCHERICHIA COLI (E. coli) / References: UniProt: Q7WT50#2: Chemical | Salicylic acid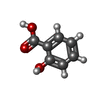  Mass: 138.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H6O3 Mass: 138.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H6O3#3: Chemical | Polyethylene glycol  Mass: 194.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, LEU 80 TO MET ENGINEERED RESIDUE IN CHAIN B, LEU 80 TO MET ...ENGINEERED | Nonpolymer details | PEG 400 (PG4): PEG 3350 IN CRYSTALLIS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47 % / Description: RD1, RD2 USED AS SEPARATE SEARCH MODELS |
|---|---|
| Crystal grow | Details: 0.1M BIS-TRIS PH 6.5, 0.2M MGCL2, 25% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.873 / Beamline: ID23-2 / Wavelength: 0.873 |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→47.1 Å / Num. obs: 20475 / % possible obs: 94.1 % / Observed criterion σ(I): 0 / Redundancy: 2.6 % / Biso Wilson estimate: 40.6 Å2 / Rmerge(I) obs: 0.17 / Net I/σ(I): 5.4 |
| Reflection shell | Resolution: 2.89→3.04 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.58 / Mean I/σ(I) obs: 1.7 / % possible all: 88.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1UTB, MOLECULE A Resolution: 2.89→47.14 Å / SU ML: 0.46 / σ(F): 1.34 / Phase error: 30.39 / Stereochemistry target values: ML Details: FOR ALL CHAINS IN THE ASYMMETRIC UNIT ELECTRON DENSITY FOR AMINO ACIDS ALA 201 TO VAL 209 IS DISORDERED. THE MODEL IN THESE REGIONS SHOULD BE TREATED WITH CAUTION.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 36.22 Å2 / ksol: 0.35 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.3 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.89→47.14 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
