 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vqz: Structure of the cap-binding domain of influenza virus polymerase... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vqz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the cap-binding domain of influenza virus polymerase subunit PB2 with bound m7GTP | ||||||
 Components Components | POLYMERASE BASIC PROTEIN 2 | ||||||
 Keywords Keywords |  TRANSCRIPTION / RNA-DEPENDENT RNA POLYMERASE / PB2 SUBUNIT / INFLUENZA VIRUS / CAP-BINDING DOMAIN TRANSCRIPTION / RNA-DEPENDENT RNA POLYMERASE / PB2 SUBUNIT / INFLUENZA VIRUS / CAP-BINDING DOMAIN | ||||||
| Function / homology |  Function and homology informationcap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / host cell mitochondrion / 7-methylguanosine mRNA capping / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / virion component / virus-mediated perturbation of host defense response / viral RNA genome replication / RNA-dependent RNA polymerase activity / DNA-templated transcription ...cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / host cell mitochondrion / 7-methylguanosine mRNA capping / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / virion component / virus-mediated perturbation of host defense response / viral RNA genome replication / RNA-dependent RNA polymerase activity / DNA-templated transcription / host cell nucleus / RNA binding Function and homology informationcap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / host cell mitochondrion / 7-methylguanosine mRNA capping / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / virion component / virus-mediated perturbation of host defense response / viral RNA genome replication / RNA-dependent RNA polymerase activity / DNA-templated transcription ...cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / host cell mitochondrion / 7-methylguanosine mRNA capping / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MAVS activity / virion component / virus-mediated perturbation of host defense response / viral RNA genome replication / RNA-dependent RNA polymerase activity / DNA-templated transcription / host cell nucleus / RNA bindingSimilarity search - Function | ||||||
| Biological species |   INFLUENZA A VIRUS INFLUENZA A VIRUS | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.3 Å | ||||||
 Authors Authors | Guilligay, D. / Tarendeau, F. / Resa-Infante, P. / Coloma, R. / Crepin, T. / Sehr, P. / Lewis, J. / Ruigrok, R.W.H. / Ortin, J. / Hart, D.J. / Cusack, S. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2008 Title: The Structural Basis for CAP Binding by Influenza Virus Polymerase Subunit Pb2. Authors: Guilligay, D. / Tarendeau, F. / Resa-Infante, P. / Coloma, R. / Crepin, T. / Sehr, P. / Lewis, J. / Ruigrok, R.W.H. / Ortin, J. / Hart, D.J. / Cusack, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vqz.cif.gz | 344.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vqz.ent.gz | 295.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2vqz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vq/2vqzftp://data.pdbj.org/pub/pdb/validation_reports/vq/2vqz | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper:
|
-Components
| #1: Protein | Mass: 19316.877 Da / Num. of mol.: 5 / Fragment: CAP-BINDING DOMAIN, RESIDUES 318-483 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) INFLUENZA A VIRUS / Strain: A/VICTORIA/3/1975(H3N2) / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21-CODONPLUS-RIL / References: UniProt: P31345 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21-CODONPLUS-RIL / References: UniProt: P31345#2: Chemical | ChemComp-MGT / 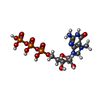  Mass: 539.223 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C11H20N5O14P3 Mass: 539.223 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C11H20N5O14P3#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 272 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 272 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, ARG 389 TO LYS ENGINEERED RESIDUE IN CHAIN B, ARG 389 TO LYS ...ENGINEERED | Nonpolymer details | 7N-METHYL-8-HYDROGUANOSINE-5'-TRIPHOSPHATE (MGT): CO-CRYSTALLISED SELENOMETHIONINE (MSE): ...7N-METHYL-8-HYDROGUANO | Sequence details | MUTATION R389K IN OUR SEQUENCE COMPARED TO DATABASE. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.51 Å3/Da / Density % sol: 51 % / Description: NONE |
|---|---|
| Crystal grow | pH: 4.6 Details: 1 MICROLITRE OF PROTEIN SOLUTION AT 11 MG PER ML IN 50 MM TRIS-HCL (PH 8.0), 200 MM NACL, 2 MM DTT, 5 MM M7GTP WITH AN EQUAL VOLUME OF A SOLUTION CONTAINING 0.1 M CITRIC ACID PH 4.6, 1.6-1.8 M SODIUM FORMATE. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 / Beamline: ID14-1 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jul 8, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→30 Å / Num. obs: 40785 / % possible obs: 98.4 % / Observed criterion σ(I): 0 / Redundancy: 3.48 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 8.59 |
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 3.38 % / Rmerge(I) obs: 0.48 / Mean I/σ(I) obs: 1.79 / % possible all: 97.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD Starting model: NONE Resolution: 2.3→29.53 Å / Cor.coef. Fo:Fc: 0.946 / Cor.coef. Fo:Fc free: 0.92 / SU B: 13.245 / SU ML: 0.16 / Cross valid method: THROUGHOUT / ESU R: 0.317 / ESU R Free: 0.227 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.47 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→29.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|