 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2tps | ||||||
|---|---|---|---|---|---|---|---|
| Title | THIAMIN PHOSPHATE SYNTHASE | ||||||
 Components Components | PROTEIN (THIAMIN PHOSPHATE SYNTHASE) | ||||||
 Keywords Keywords | THIAMIN BIOSYNTHESIS /  TIM BARREL TIM BARREL | ||||||
| Function / homology |  Function and homology information Function and homology informationthiamine phosphate synthase / thiamine-phosphate diphosphorylase activity / thiamine diphosphate biosynthetic process / thiamine biosynthetic process / magnesium ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Bacillus subtilis (bacteria) Bacillus subtilis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.25 Å | ||||||
 Authors Authors | Chiu, H.-J. / Reddick, J.J. / Begley, T.P. / Ealick, S.E. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Crystal structure of thiamin phosphate synthase from Bacillus subtilis at 1.25 A resolution. Authors: Chiu, H.J. / Reddick, J.J. / Begley, T.P. / Ealick, S.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2tps.cif.gz | 165.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2tps.ent.gz | 126.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2tps.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tp/2tpsftp://data.pdbj.org/pub/pdb/validation_reports/tp/2tps | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 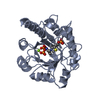
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.999757, -0.021512, 0.004717), Vector : |
-Components
| #1: Protein | Mass: 24312.643 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Bacillus subtilis (bacteria) / Strain: DE3 / References: UniProt: P39594, thiamine phosphate synthase#2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 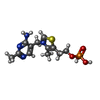  Mass: 345.334 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O4PS Mass: 345.334 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O4PS#4: Chemical | Pyrophosphate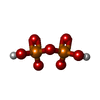  Mass: 175.959 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: H2O7P2 Mass: 175.959 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: H2O7P2#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 444 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 444 / Source method: isolated from a natural source / Formula: H2OSequence details | THE FOLLOWING RESIDUES ARE NOT PART OF THE SWISS-PROT DATABASE ENTRY FOR CHAINS A & B: HIS9, HIS10, ...THE FOLLOWING RESIDUES ARE NOT PART OF THE SWISS-PROT DATABASE ENTRY FOR CHAINS A & B: HIS9, HIS10, GLY11, ILE12, ARG13. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.05 Å3/Da / Density % sol: 35 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: CRYSTALLIZATION CONDITIONS: 75 MM TRIS-HCL, PH 7.5 21-22% PEG4000 75 MM MGCL2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.9795,0.9792,0.9677 / Beamline: F1 / Wavelength: 0.9795,0.9792,0.9677 | ||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD | ||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 1.25→30 Å / Num. obs: 94169 / % possible obs: 81.6 % / Redundancy: 5.1 % / Rsym value: 0.062 / Net I/σ(I): 6.8 | ||||||||||||
| Reflection shell | Resolution: 1.25→1.32 Å / Redundancy: 3 % / Mean I/σ(I) obs: 3.4 / Rsym value: 0.219 / % possible all: 48.5 | ||||||||||||
| Reflection | *PLUS Rmerge(I) obs: 0.062 | ||||||||||||
| Reflection shell | *PLUS % possible obs: 48.5 % / Rmerge(I) obs: 0.219 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.25→8 Å / Num. parameters: 18915 / Num. restraintsaints: 18840 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: THE STRUCTURE WAS REFINED WITH X-PLOR AND FOLLOWED BY SHELXL
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 9 / Occupancy sum non hydrogen: 3962.6 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.25→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
