 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2tod: ORNITHINE DECARBOXYLASE FROM TRYPANOSOMA BRUCEI K69A MUTANT IN CO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2tod | ||||||
|---|---|---|---|---|---|---|---|
| Title | ORNITHINE DECARBOXYLASE FROM TRYPANOSOMA BRUCEI K69A MUTANT IN COMPLEX WITH ALPHA-DIFLUOROMETHYLORNITHINE | ||||||
 Components Components | PROTEIN (ORNITHINE DECARBOXYLASE) | ||||||
 Keywords Keywords |  LYASE / POLYAMINE METABOLISM / PYRIDOXAL 5'-PHOSPHATE / ALPHA-BETA BARREL LYASE / POLYAMINE METABOLISM / PYRIDOXAL 5'-PHOSPHATE / ALPHA-BETA BARREL | ||||||
| Function / homology |  Function and homology informationornithine decarboxylase / putrescine biosynthetic process from ornithine / ornithine decarboxylase activity / polyamine biosynthetic process Function and homology informationornithine decarboxylase / putrescine biosynthetic process from ornithine / ornithine decarboxylase activity / polyamine biosynthetic processSimilarity search - Function | ||||||
| Biological species |  Trypanosoma brucei (eukaryote) Trypanosoma brucei (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Grishin, N.V. / Osterman, A.L. / Brooks, H.B. / Phillips, M.A. / Goldsmith, E.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with alpha-difluoromethylornithine. Authors: Grishin, N.V. / Osterman, A.L. / Brooks, H.B. / Phillips, M.A. / Goldsmith, E.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2tod.cif.gz | 305.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2tod.ent.gz | 243.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2tod.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/to/2todftp://data.pdbj.org/pub/pdb/validation_reports/to/2tod | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qu4C  7odcS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 47004.359 Da / Num. of mol.: 4 / Mutation: K69A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Trypanosoma brucei (eukaryote) / Cellular location (production host): CYTOPLASM / Production host:  Escherichia coli (E. coli) / Strain (production host): B21/DG3 / References: UniProt: P07805, ornithine decarboxylase Escherichia coli (E. coli) / Strain (production host): B21/DG3 / References: UniProt: P07805, ornithine decarboxylase#2: Chemical | ChemComp-DMO / Eflornithine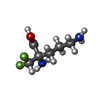  Mass: 182.168 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H12F2N2O2 / Comment: medication*YM Mass: 182.168 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H12F2N2O2 / Comment: medication*YM#3: Chemical | ChemComp-PLP / Pyridoxal phosphate  Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H10NO6P#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 869 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 869 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 32 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: 16% PEG 3350, 0.2 M SOLIUM ACETATE, 100MM HEPES PH 7.5, 20MG/ML PROTEIN DILUTED TWICE, 16C, pH 7.0 | ||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 16 ℃ / pH: 8 / Method: vapor diffusion, hanging dropDetails: Grishin, N.V., (1996) Proteins Struct. Funct. Genet., 24, 272. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.908 / Beamline: A1 / Wavelength: 0.908 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.908 Å / Relative weight: 1 |
| Reflection | Resolution: 2→40 Å / Num. obs: 103282 / % possible obs: 97.6 % / Redundancy: 3.3 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 13.9 |
| Reflection | *PLUS Num. measured all: 339850 |
| Reflection shell | *PLUS % possible obs: 61.7 % / Mean I/σ(I) obs: 1.9 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 7ODC Resolution: 2→8 Å / Cross valid method: THROUGHOUT / σ(F): 1
| ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→8 Å
| ||||||||||||||||
| Software | *PLUS Name: 'REFMAC, X-PLOR, CNS' / Classification: refinement | ||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / σ(F): 1 / % reflection Rfree: 10 % / Rfactor obs: 0.212 / Rfactor Rfree: 0.27 | ||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||
| Refine LS restraints | *PLUS
|
