 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2oqq: Crystal structure of HY5 leucine zipper homodimer from Arabidopsi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2oqq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of HY5 leucine zipper homodimer from Arabidopsis thaliana | ||||||
 Components Components | Transcription factor HY5 | ||||||
 Keywords Keywords |  TRANSCRIPTION / homodimer leucine zipper TRANSCRIPTION / homodimer leucine zipper | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of anthocyanin metabolic process / red or far-red light signaling pathway / regulation of photomorphogenesis / regulation of abscisic acid-activated signaling pathway / response to red light / positive regulation of flavonoid biosynthetic process / positive gravitropism / gibberellic acid mediated signaling pathway / red, far-red light phototransduction / response to far red light ...positive regulation of anthocyanin metabolic process / red or far-red light signaling pathway / regulation of photomorphogenesis / regulation of abscisic acid-activated signaling pathway / response to red light / positive regulation of flavonoid biosynthetic process / positive gravitropism / gibberellic acid mediated signaling pathway / red, far-red light phototransduction / response to far red light / response to abscisic acid / positive regulation of circadian rhythm / abscisic acid-activated signaling pathway / response to UV-B / response to cold / protein-DNA complex / double-stranded DNA binding / transcription cis-regulatory region binding / DNA-binding transcription factor activity, RNA polymerase II-specific / DNA-binding transcription factor activity / positive regulation of transcription by RNA polymerase II / DNA binding / nucleusSimilarity search - Function | ||||||
| Biological species |  Arabidopsis thaliana (thale cress) Arabidopsis thaliana (thale cress) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2 Å | ||||||
 Authors Authors | Yoon, M.-K. / Kim, H.M. / Choi, G. / Lee, J.-O. / Choi, B.-S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: Structural basis for the conformational integrity of the Arabidopsis thaliana HY5 leucine zipper homodimer. Authors: Yoon, M.K. / Kim, H.M. / Choi, G. / Lee, J.O. / Choi, B.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2oqq.cif.gz | 31.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2oqq.ent.gz | 20.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2oqq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oq/2oqqftp://data.pdbj.org/pub/pdb/validation_reports/oq/2oqq | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 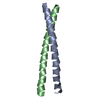
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 4964.506 Da / Num. of mol.: 2 / Fragment: Hy5 leucine zipper Source method: isolated from a genetically manipulated source Source: (gene. exp.) Arabidopsis thaliana (thale cress) / Plasmid: pGEX4T3 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: O24646 Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: O24646#2: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 102 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45.45 % |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 0.1M Bis-Tris pH6.0 0.1M MgCl2 25% PEG 3350, VAPOR DIFFUSION, HANGING DROP, temperature 296K |
-Data collection
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS  / Beamline: 6B / Wavelength: 1.12714, 0.97880, 0.97895, 0.97114 / Beamline: 6B / Wavelength: 1.12714, 0.97880, 0.97895, 0.97114 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 6080 / % possible obs: 97.7 % / Redundancy: 2.9 % / Rmerge(I) obs: 0.095 / Net I/σ(I): 9.1 | |||||||||||||||
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.392 / Num. unique all: 553 / % possible all: 87.8 |
-Phasing
| Phasing | Method: SIRAS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing dm | FOM : 0.77 / FOM acentric: 0.78 / FOM centric: 0.74 / Reflection: 5190 / Reflection acentric: 4648 / Reflection centric: 542 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MIR | Resolution: 2→20 Å / FOM: 0.595 / FOM acentric: 0.598 / FOM centric: 0.571 / Reflection: 5190 / Reflection acentric: 4648 / Reflection centric: 542 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MIR der | Der set-ID: 1
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MIR der shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MIR der site | Biso : 25 Å / Atom type symbol: Au
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MIR shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS / Resolution: 2→20 Å / σ(F): 0
| ||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 47.692 Å2 | ||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.094 Å2
| ||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→20 Å
| ||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||
| Xplor file |
|
