 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2o3z: X-ray crystal structure of LpxC complexed with 3-heptyloxybenzoate -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2o3z | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray crystal structure of LpxC complexed with 3-heptyloxybenzoate | ||||||
 Components Components | UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine deacetylase | ||||||
 Keywords Keywords |  HYDROLASE / Lipid A biosynthesis / Lipid synthesis / LpxC / 3-heptyloxybenzoate HYDROLASE / Lipid A biosynthesis / Lipid synthesis / LpxC / 3-heptyloxybenzoate | ||||||
| Function / homology |  Function and homology informationUDP-3-O-acyl-N-acetylglucosamine deacetylase / UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine deacetylase activity / UDP-3-O-acyl-N-acetylglucosamine deacetylase activity / lipid A biosynthetic process / metal ion binding Function and homology informationUDP-3-O-acyl-N-acetylglucosamine deacetylase / UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine deacetylase activity / UDP-3-O-acyl-N-acetylglucosamine deacetylase activity / lipid A biosynthetic process / metal ion bindingSimilarity search - Function | ||||||
| Biological species |   Aquifex aeolicus (bacteria) Aquifex aeolicus (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Gennadios, H.A. / Whittington, D.A. / Christianson, D.W. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem. / Year: 2007 Title: Amphipathic benzoic acid derivatives: synthesis and binding in the hydrophobic tunnel of the zinc deacetylase LpxC. Authors: Shin, H. / Gennadios, H.A. / Whittington, D.A. / Christianson, D.W. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE THE RESIDUE NUMBERING SYSTEM FOLLOWS THAT OF THE E.COLI ENZYME, AND RESULTS IN A BREAKS IN ...SEQUENCE THE RESIDUE NUMBERING SYSTEM FOLLOWS THAT OF THE E.COLI ENZYME, AND RESULTS IN A BREAKS IN THE SEQUENTIAL NUMBERING IN FEW REGIONS |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2o3z.cif.gz | 125.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2o3z.ent.gz | 97.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2o3z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o3/2o3zftp://data.pdbj.org/pub/pdb/validation_reports/o3/2o3z | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1p42S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | There are 2 monomers in the asymmetric unit forming a crystallograpic dimer. The second monomer is generated by: X,Y,Z Y,X-Y,1/3+Z -X+Y,-X,2/3+Z -X,-Y,1/2+Z Y,-X+Y,5/6+Z X-Y,X,1/6+Z |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 30989.510 Da / Num. of mol.: 2 / Mutation: deltaD284-L294, C193A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Aquifex aeolicus (bacteria) / Gene: lpxC, envA / Plasmid: pET21a / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3)pLysSReferences: UniProt: O67648, Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amides |
|---|
-Non-polymers , 5 types, 178 molecules 


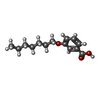

| #2: Chemical | Sulfate Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-CL / | Chloride Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#5: Chemical |  Mass: 236.307 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20O3 Mass: 236.307 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20O3#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 168 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 57.79 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 100mM HEPES pH 7.0, 12-14% PEG3350, 180mM NaCl, 0.5mM ZnSO4, VAPOR DIFFUSION, HANGING DROP, temperature 294K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X29A / Wavelength: 0.9 Å / Beamline: X29A / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Jun 6, 2005 Details: sagital focusing monochromator and vertical focusing mirror |
| Radiation | Monochromator: Rosenbaum-Rock double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 2.25→50 Å / Num. obs: 33203 / % possible obs: 98.7 % / Redundancy: 2.8 % / Biso Wilson estimate: 24.6 Å2 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 2.25→2.28 Å / % possible obs: 99.8 % / Redundancy: 2.6 % / Mean I/σ(I) obs: 2.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1P42 Resolution: 2.25→46.7 Å / Cross valid method: THROUGHOUT / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å / Luzzati d res low obs: 5 Å / Luzzati sigma a obs: 0.21 Å | ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→46.7 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell |
|
