1 average structure + 20 structures with the lowest energy
Representative
Model #1
average structure
-
Components
#1: Protein/peptide
Thymosinalpha-1
Mass: 3096.307 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: The protein was made on a protein synthesizer. / Source: (synth.) Homo sapiens (human) / References: UniProt: P06454
Case, Darden, Cheatham, III, Simmerling, Wang, Duke, Luo, andKollm
moleculardynamics
AMBER
9
Case, Darden, Cheatham, III, Simmerling, Wang, Duke, Luo, andKollm
minimization
AMBER
9
Case, Darden, Cheatham, III, Simmerling, Wang, Duke, Luo, andKollm
dataanalysis
AMBER
refinement
Refinement
Method: molecular dynamics, restrained molecular dynamics, restrained molecular dynamics, selection, average structure Software ordinal: 1 Details: 25 ns at 300K on a explicit solvent ratio 40% TFE/H2O (v/v), with a linear-helix initial structure, After the 25 ns on explicit solvent, 1 ns GB simulation follows with all (549)restrains at ...Details: 25 ns at 300K on a explicit solvent ratio 40% TFE/H2O (v/v), with a linear-helix initial structure, After the 25 ns on explicit solvent, 1 ns GB simulation follows with all (549)restrains at 300K., After the 1ns GB restrained simulation, a 10ns restrained MD simulation follows on a explicit solvent ratio 40% TFE/water (v/v). 1000 structures were collected (1 every 10ps), minimized and sorted by total energy. The best 20 structures with the lowest energy were selected., The average structure was obtained from the average of the 20 best structures with the lowest energy and minimized with the 549 restraints and the GB method in order to taking in account the salvation implicitly.
NMR constraints
NOE constraints total: 415 / NOE intraresidue total count: 170 / NOE long range total count: 0 / NOE medium range total count: 106 / NOE sequential total count: 139 / Protein chi angle constraints total count: 0 / Protein other angle constraints total count: 100 / Protein phi angle constraints total count: 17 / Protein psi angle constraints total count: 17
NMR representative
Selection criteria: average structure
NMR ensemble
Average torsion angle constraint violation: 0.075 ° Conformer selection criteria: 1 average structure + 20 structures with the lowest energy Conformers calculated total number: 1000 / Conformers submitted total number: 21 / Maximum lower distance constraint violation: 0.033 Å / Maximum torsion angle constraint violation: 4.081 ° / Maximum upper distance constraint violation: 0.368 Å Torsion angle constraint violation method: minimizing the sum of the absolute values of the errors
NMR ensemble rms
Distance rms dev: 0.0086 Å / Distance rms dev error: 0.036 Å
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSCRIPTION
TRANSCRIPTION Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads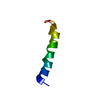






 PDBj
PDBj Assembly
Assembly
 Sample preparation
Sample preparation Processing
Processing