 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ihu: Carboxyethylarginine synthase from Streptomyces clavuligerus: put... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ihu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Carboxyethylarginine synthase from Streptomyces clavuligerus: putative reaction intermediate complex | ||||||
 Components Components | Carboxyethylarginine synthase | ||||||
 Keywords Keywords |  TRANSFERASE / Thiamin diphosphate complex TRANSFERASE / Thiamin diphosphate complex | ||||||
| Function / homology |  Function and homology informationN2-(2-carboxyethyl)arginine synthase / N2-(2-carboxyethyl)arginine synthase activity / thiamine pyrophosphate binding / magnesium ion binding Function and homology informationN2-(2-carboxyethyl)arginine synthase / N2-(2-carboxyethyl)arginine synthase activity / thiamine pyrophosphate binding / magnesium ion bindingSimilarity search - Function | ||||||
| Biological species |  Streptomyces clavuligerus (bacteria) Streptomyces clavuligerus (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Caines, M.E. / Schofield, C.J. | ||||||
 Citation Citation | Journal: Biochem.Biophys.Res.Commun. / Year: 2009 Title: Structural and mechanistic studies on N(2)-(2-carboxyethyl)arginine synthase. Authors: Caines, M.E. / Sorensen, J.L. / Schofield, C.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ihu.cif.gz | 454 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ihu.ent.gz | 365.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2ihu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ih/2ihuftp://data.pdbj.org/pub/pdb/validation_reports/ih/2ihu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ihtC  2ihvC  1upaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: LYS / Beg label comp-ID: LYS / Refine code: 4
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 60964.828 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces clavuligerus (bacteria) / Gene: ceas2 / Plasmid: pET24a(+) / Species (production host): Escherichia coli / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3)References: UniProt: Q9LCV9, N2-(2-carboxyethyl)arginine synthase |
|---|
-Non-polymers , 7 types, 1423 molecules 


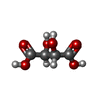



| #2: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: K#4: Chemical | ChemComp-TP9 / (  Mass: 412.296 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H18N4O7P2S Mass: 412.296 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H18N4O7P2S#5: Chemical | Tartaric acid Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6#6: Chemical | ChemComp-GOL / | Glycerol Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#7: Chemical | ChemComp-TP8 / |  Mass: 574.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21N4O12P3S Mass: 574.333 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21N4O12P3S#8: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 1405 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.09 Å3/Da / Density % sol: 60.21 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: 1.6 M (NH4)2SO4, 0.1 M HEPES pH 7.4, 10 mg/mL protein with 3-fold excess of ThDP, VAPOR DIFFUSION, HANGING DROP, temperature 290K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.813 Å / Beamline: X11 / Wavelength: 0.813 Å |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Dec 19, 2004 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.813 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→28.133 Å / Num. all: 189093 / Num. obs: 183609 / % possible obs: 97.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.7 % / Biso Wilson estimate: 20.17 Å2 / Rmerge(I) obs: 0.079 / Rsym value: 0.079 / Net I/σ(I): 7.9 |
| Reflection shell | Resolution: 2.05→2.16 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.305 / Mean I/σ(I) obs: 2.4 / Rsym value: 0.305 / % possible all: 88.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1UPA Resolution: 2.05→28.13 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.939 / SU B: 3.457 / SU ML: 0.094 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.152 / ESU R Free: 0.136 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: This data was collected on a crystal of CEAS soaked with 3 mM DL-glyceraldehyde-3-phosphate, in a solution containing 2 M dipotassium tartrate. The electron density in the active sites of ...Details: This data was collected on a crystal of CEAS soaked with 3 mM DL-glyceraldehyde-3-phosphate, in a solution containing 2 M dipotassium tartrate. The electron density in the active sites of subunits A and D could be interpreted as either tartrate or glyceraldehyde-3-phosphate, or as a mixture. The model has been built conservatively with tartrate bound. The electron density in the active site of subunit C appeared consistent with the formation of a covalently bound species and a number of mechanistically plausible ligands were modelled. Of the potential structures tested, the enol(ate)-ThDP species was judged most consistent with the experimental data, however other possibilities exist and the model should be treated with due caution.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.712 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.05→28.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 4033 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.05→2.103 Å / Total num. of bins used: 20
|
