 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2i2q | ||||||
|---|---|---|---|---|---|---|---|
| Title | Fission Yeast cofilin | ||||||
 Components Components | Cofilin ADF/Cofilin family ADF/Cofilin family | ||||||
 Keywords Keywords | actin-binding protein / N-terminal Serine | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of actin filament binding / actin filament debranching / mitotic actomyosin contractile ring / medial cortex / mitotic actomyosin contractile ring assembly / actin nucleation / cell tip / actin cortical patch / actin filament severing / actin filament depolymerization ...negative regulation of actin filament binding / actin filament debranching / mitotic actomyosin contractile ring / medial cortex / mitotic actomyosin contractile ring assembly / actin nucleation / cell tip / actin cortical patch / actin filament severing / actin filament depolymerization / cell division site / actin monomer binding / nuclear matrix / actin filament binding / actin cytoskeleton / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Schizosaccharomyces pombe (fission yeast) Schizosaccharomyces pombe (fission yeast) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.72 Å | ||||||
 Authors Authors | Andrianantoandro, E. / Pollard, T.D. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 2006 Title: Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Authors: Andrianantoandro, E. / Pollard, T.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2i2q.cif.gz | 41.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2i2q.ent.gz | 27.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2i2q.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i2/2i2qftp://data.pdbj.org/pub/pdb/validation_reports/i2/2i2q | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1cofS S: Starting model for refinement |
|---|---|
| Similar structure data |







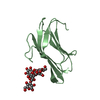


-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | ADF/Cofilin family Mass: 15640.864 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Schizosaccharomyces pombe (fission yeast)Gene: cof1 / Production host:  Escherichia coli (E. coli) / References: UniProt: P78929 Escherichia coli (E. coli) / References: UniProt: P78929 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-LDA / | Lauryldimethylamine oxide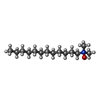  Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM#4: Chemical | ChemComp-EDO / Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 90 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 90 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.32 Å3/Da / Density % sol: 64 % |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 2.1 M Ammonium Sulfate, 100 mM imidazole pH 7.0, 1 mM NiSO4, 2.5% PEG 400, 1 mM LDAO, VAPOR DIFFUSION, SITTING DROP, temperature 296K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: May 26, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→50 Å / Num. obs: 25210 / % possible obs: 88.8 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Rmerge(I) obs: 0.071 / Χ2: 5.212 / Net I/σ(I): 18.5 |
| Reflection shell | Resolution: 1.6→1.66 Å / Num. unique all: 981 / Χ2: 7.956 / % possible all: 35.4 |
-Phasing
| Phasing MR | Cor.coef. Fo:Fc: 0.499 / Packing: 0.381
|
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1COF Resolution: 1.72→30 Å / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 52.597 Å2 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.406 Å2
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.72→30 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
