 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cof | ||||||
|---|---|---|---|---|---|---|---|
| Title | YEAST COFILIN, ORTHORHOMBIC CRYSTAL FORM | ||||||
 Components Components | COFILIN ADF/Cofilin family ADF/Cofilin family | ||||||
 Keywords Keywords | ACTIN-BINDING PROTEIN / ACTIN-BINDING / CYTOSKELETON | ||||||
| Function / homology |  Function and homology information Function and homology informationactin cortical patch / Golgi to plasma membrane protein transport / actin filament severing / actin filament depolymerization / actin filament organization / nuclear matrix / endocytosis / actin filament binding / actin cytoskeleton / plasma membrane / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / MIR / Resolution: 2.3 Å | ||||||
 Authors Authors | Fedorov, A.A. / Lappalainen, P. / Fedorov, E.V. / Drubin, D.G. / Almo, S.C. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1997 Title: Structure determination of yeast cofilin. Authors: Fedorov, A.A. / Lappalainen, P. / Fedorov, E.V. / Drubin, D.G. / Almo, S.C. #1: Journal: Mol.Cell.Biol. / Year: 1995Title: The Adf/Cofilin Proteins: Stimulus-Responsive Modulators of Actin Dynamics Authors: Moon, A. / Drubin, D.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
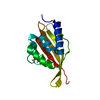
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cof.cif.gz | 37.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cof.ent.gz | 26.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1cof.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/co/1cofftp://data.pdbj.org/pub/pdb/validation_reports/co/1cof | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | ADF/Cofilin family Mass: 15919.742 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Production host:  Escherichia coli (E. coli) / References: UniProt: Q03048 Escherichia coli (E. coli) / References: UniProt: Q03048 |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 38 % |
|---|---|
| Crystal grow | pH: 7.2 / Details: CRYSTALLIZED FROM 25% PEG 4000 SOLUTION., pH 7.2 |
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / PH range low: 7.5 / PH range high: 7 |
| Components of the solutions | *PLUS Conc.: 20-30 % / Common name: PEG4000 |
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: 1995 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→26.2 Å / Num. obs: 6330 / % possible obs: 87.5 % / Observed criterion σ(I): 2 / Redundancy: 2.33 % / Rmerge(I) obs: 0.049 / Net I/σ(I): 18.5 |
| Reflection | *PLUS Rmerge(I) obs: 0.067 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 2.3→8 Å / σ(F): 2 Details: N-TERMINAL RESIDUES 1 - 5 AND C-TERMINAL RESIDUES 141 - 143 ARE NOT VISIBLE IN THE MAPS. LOOP 73 - 78 HAS WEAK ELECTRON DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.23 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
