 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2f2g: X-Ray Structure of Gene Product From Arabidopsis Thaliana AT3G16990 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2f2g | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | X-Ray Structure of Gene Product From Arabidopsis Thaliana AT3G16990 | |||||||||
 Components Components | SEED MATURATION PROTEIN PM36 HOMOLOG | |||||||||
 Keywords Keywords |  PLANT PROTEIN / TENA_THI-4 DOMAIN / TENA/THI-4/PQQC FAMILY / AT3G16990 / STRUCTURAL GENOMICS / PROTEIN STRUCTURE INITIATIVE / PSI / CESG / CENTER FOREUKARYOTIC STRUCTURAL GENOMICS / Center for Eukaryotic Structural Genomics PLANT PROTEIN / TENA_THI-4 DOMAIN / TENA/THI-4/PQQC FAMILY / AT3G16990 / STRUCTURAL GENOMICS / PROTEIN STRUCTURE INITIATIVE / PSI / CESG / CENTER FOREUKARYOTIC STRUCTURAL GENOMICS / Center for Eukaryotic Structural Genomics | |||||||||
| Function / homology |  Function and homology informationaminopyrimidine aminohydrolase / thiaminase activity / thiamine diphosphate biosynthetic process / thiamine biosynthetic process / Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amides Function and homology informationaminopyrimidine aminohydrolase / thiaminase activity / thiamine diphosphate biosynthetic process / thiamine biosynthetic process / Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amidesSimilarity search - Function | |||||||||
| Biological species |  Arabidopsis thaliana (thale cress) Arabidopsis thaliana (thale cress) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / MAD / Resolution: 2.1 Å | |||||||||
 Authors Authors | Wesenberg, G.W. / Smith, D.W. / Phillips Jr., G.N. / Johnson, K.A. / Bitto, E. / Bingman, C.A. / Center for Eukaryotic Structural Genomics (CESG) | |||||||||
 Citation Citation | Journal: Proteins / Year: 2004 Title: Crystal structure of gene locus At3g16990 from Arabidopsis thaliana Authors: Blommel, P.G. / Smith, D.W. / Bingman, C.A. / Dyer, D.H. / Rayment, I. / Holden, H.M. / Fox, B.G. / Phillips Jr., G.N. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2f2g.cif.gz | 106.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2f2g.ent.gz | 86.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2f2g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f2/2f2gftp://data.pdbj.org/pub/pdb/validation_reports/f2/2f2g | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / Refine code: 4
|
-Components
| #1: Protein | Mass: 25332.719 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Arabidopsis thaliana (thale cress) / Gene: At3g16990 / Plasmid: PVP13 / Production host:  Escherichia coli (E. coli) / Strain (production host): Rosetta / References: UniProt: Q9ASY9 Escherichia coli (E. coli) / Strain (production host): Rosetta / References: UniProt: Q9ASY9#2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 4-Amino-5-hydroxymethyl-2-methylpyrimidine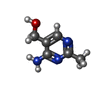  Mass: 139.155 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H9N3O Mass: 139.155 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H9N3O#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.5 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop Details: Protein solution (15 mg/ml protein, 0.025 M sodium chloride, 0.10 M Tris pH 7.5) mixed in a 1:1 ratio with the well solution (0.055 M sodium acetate pH 4.5, 1.05 M ammonium sulfate), ...Details: Protein solution (15 mg/ml protein, 0.025 M sodium chloride, 0.10 M Tris pH 7.5) mixed in a 1:1 ratio with the well solution (0.055 M sodium acetate pH 4.5, 1.05 M ammonium sulfate), temperature 277 K, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 90 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-ID-B / Wavelength: 0.97912 Å / Beamline: 14-ID-B / Wavelength: 0.97912 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jun 26, 2003 / Details: Bent cylindrical Si-mirror (Rh coating) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Diamond (111) double-crystal monochromator / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97912 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.08→43.81 Å / Num. obs: 30515 / % possible obs: 84.9 % / Redundancy: 9.5 % / Rmerge(I) obs: 0.065 / Net I/σ(I): 22.303 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: MAD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MAD | D res high: 2.5 Å / D res low: 25 Å / FOM : 0.47 / Reflection: 20370 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set site |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm | FOM : 0.69 / FOM acentric: 0.73 / FOM centric: 0.5 / Reflection: 20370 / Reflection acentric: 16709 / Reflection centric: 3661 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.1→43.81 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.922 / SU B: 8.176 / SU ML: 0.114 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.209 / ESU R Free: 0.194 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS, SELENIUM C COEFFICIENT FOR STRUCTURE FACTOR CALCULATION SET TO -9.00, MOLPROBITY USED TO ASSIST IN FINAL MODEL BUILDING.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.599 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→43.81 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 1452 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.101→2.156 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
