Entry Database : PDB / ID : 1usbTitle Rational design of a novel enzyme - efficient thioester hydrolysis enabled by the incorporation of a single His residue into human glutathione transferase A1-1 GLUTATHIONE S-TRANSFERASE A1 Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species HOMO SAPIENS (human)Method / / / Resolution : 2.07 Å Authors Jakobsson, E. / Kleywegt, G.J. Journal : Proc.Natl.Acad.Sci.USA / Year : 2004Title : Incorporation of a Single His Residue by Rational Design Enables Thiol-Ester Hydrolysis by Human Glutathione Transferase A1-1Authors : Hederos, S. / Broo, K.S. / Jakobsson, E. / Kleywegt, G.J. / Mannervik, B. / Baltzer, L. History Deposition Nov 20, 2003 Deposition site / Processing site Revision 1.0 Sep 1, 2004 Provider / Type Revision 1.1 May 8, 2011 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Jan 17, 2018 Group / Data collection / Category / pdbx_unobs_or_zero_occ_atoms / Item Revision 1.4 Oct 9, 2019 Group / Experimental preparation / Other / Category / pdbx_database_statusItem / _pdbx_database_status.status_code_sfRevision 1.5 Dec 13, 2023 Group Advisory / Data collection ... Advisory / Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / pdbx_unobs_or_zero_occ_atoms / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components
 Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

















 PDBj
PDBj



 Assembly
Assembly

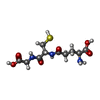
 Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
 Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
 Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 18.015 Da / Num. of mol.: 259 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 259 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I711 / Wavelength: 1.007
/ Beamline: I711 / Wavelength: 1.007  Processing
Processing