 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1k3l: Crystal Structure Analysis of S-hexyl-glutathione Complex of Glut... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1k3l | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure Analysis of S-hexyl-glutathione Complex of Glutathione Transferase at 1.5 Angstroms Resolution | ||||||
 Components Components | GLUTATHIONE S-TRANSFERASE A1 | ||||||
 Keywords Keywords | TRANSFERASE / glutathione S-transferase / S-hexyl glutathione / water structure | ||||||
| Function / homology |  Function and homology informationIsomerases; Intramolecular oxidoreductases; Transposing C=C bonds / glutathione derivative biosynthetic process / steroid delta-isomerase activity / linoleic acid metabolic process / Glutathione conjugation / glutathione peroxidase activity / Azathioprine ADME / Heme degradation / NFE2L2 regulating anti-oxidant/detoxification enzymes / prostaglandin metabolic process ...Isomerases; Intramolecular oxidoreductases; Transposing C=C bonds / glutathione derivative biosynthetic process / steroid delta-isomerase activity / linoleic acid metabolic process / Glutathione conjugation / glutathione peroxidase activity / Azathioprine ADME / Heme degradation / NFE2L2 regulating anti-oxidant/detoxification enzymes / prostaglandin metabolic process / glutathione transferase / glutathione transferase activity / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / glutathione metabolic process / epithelial cell differentiation / xenobiotic metabolic process / fatty acid binding / extracellular exosome / cytosol Function and homology informationIsomerases; Intramolecular oxidoreductases; Transposing C=C bonds / glutathione derivative biosynthetic process / steroid delta-isomerase activity / linoleic acid metabolic process / Glutathione conjugation / glutathione peroxidase activity / Azathioprine ADME / Heme degradation / NFE2L2 regulating anti-oxidant/detoxification enzymes / prostaglandin metabolic process ...Isomerases; Intramolecular oxidoreductases; Transposing C=C bonds / glutathione derivative biosynthetic process / steroid delta-isomerase activity / linoleic acid metabolic process / Glutathione conjugation / glutathione peroxidase activity / Azathioprine ADME / Heme degradation / NFE2L2 regulating anti-oxidant/detoxification enzymes / prostaglandin metabolic process / glutathione transferase / glutathione transferase activity / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / glutathione metabolic process / epithelial cell differentiation / xenobiotic metabolic process / fatty acid binding / extracellular exosome / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Le Trong, I. / Stenkamp, R.E. / Ibarra, C. / Atkins, W.M. / Adman, E.T. | ||||||
 Citation Citation | Journal: Proteins / Year: 2002 Title: 1.3-A resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site Authors: Le Trong, I. / Stenkamp, R.E. / Ibarra, C. / Atkins, W.M. / Adman, E.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1k3l.cif.gz | 211.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1k3l.ent.gz | 169.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1k3l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k3/1k3lftp://data.pdbj.org/pub/pdb/validation_reports/k3/1k3l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1k3oC  1k3yC  1gseS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / GSTA1-1 Mass: 25538.924 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  Escherichia coli (E. coli) / References: UniProt: P08263, glutathione transferase Escherichia coli (E. coli) / References: UniProt: P08263, glutathione transferase#2: Chemical | 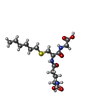  Mass: 392.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H30N3O6S Mass: 392.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H30N3O6S#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 477 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 477 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.04 Å3/Da / Density % sol: 39.8 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 18% PEG3350, 0.1 M Tris-Cl pH 7.5, 10mM DTT, VAPOR DIFFUSION, SITTING DROP, temperature 294K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 / Beamline: BL9-1 / Wavelength: 0.98 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 15, 1999 / Details: mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→40 Å / Num. all: 75496 / Num. obs: 75496 / % possible obs: 99.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rmerge(I) obs: 0.048 |
| Reflection shell | Resolution: 1.5→1.55 Å / Rmerge(I) obs: 0.444 / % possible all: 94.2 |
| Reflection | *PLUS Rmerge(I) obs: 0.048 |
| Reflection shell | *PLUS % possible obs: 94.2 % / Num. unique obs: 7153 / Rmerge(I) obs: 0.44 / Mean I/σ(I) obs: 2.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GSE [LIGANDS AND WATER REMOVED] Resolution: 1.5→10 Å / Num. parameters: 37081 / Num. restraintsaints: 44934 / Cross valid method: FREE R / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 3 / Occupancy sum hydrogen: 3725 / Occupancy sum non hydrogen: 4094 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.5 Å / Lowest resolution: 10 Å / % reflection Rfree: 10 % / Rfactor all: 0.1654 / Rfactor Rfree: 0.219 / Rfactor Rwork: 0.136 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.241 / Rfactor Rwork: 0.158 |
