 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nyb: SOLUTION STRUCTURE OF THE BACTERIOPHAGE PHI21 N PEPTIDE-BOXB RNA ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nyb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | SOLUTION STRUCTURE OF THE BACTERIOPHAGE PHI21 N PEPTIDE-BOXB RNA COMPLEX | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSCRIPTION/RNA / peptide-rna complex / transcription antitermination / TRANSCRIPTION-RNA COMPLEX | |||||||||
| Function / homology | DNA-templated transcription termination /  RNA / RNA (> 10) / Probable regulatory protein N RNA / RNA (> 10) / Probable regulatory protein N Function and homology information Function and homology information | |||||||||
| Biological species |  Enterobacteria phage phi21 (virus) Enterobacteria phage phi21 (virus) | |||||||||
| Method | SOLUTION NMR | |||||||||
| Model type details | minimized average | |||||||||
 Authors Authors | Cilley, C.D. / Williamson, J.R. | |||||||||
 Citation Citation | Journal: RNA / Year: 2003 Title: Structural mimicry in the phage phi21 N peptide-boxB RNA complex Authors: Cilley, C.D. / Williamson, J.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nyb.cif.gz | 341.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nyb.ent.gz | 295.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1nyb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ny/1nybftp://data.pdbj.org/pub/pdb/validation_reports/ny/1nyb | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 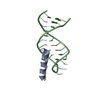
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: RNA chain | Mass: 7660.590 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: Synthesis of the RNA fragment from the Bacteriophage phi21 boxB |
|---|---|
| #2: Protein/peptide | Mass: 2584.977 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage phi21 (virus) / Gene: N / Production host:  Escherichia coli (E. coli) / References: UniProt: P07243 Escherichia coli (E. coli) / References: UniProt: P07243 |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR details | Text: The structure was determined using 2D homonuclear and 3D heteronuclear NMR spectroscopy. The two most important samples were uniformly labeled 15N,13C peptide or RNA in complex with its unlabeled counterpart. |
- Sample preparation
Sample preparation
| Details | Contents: 2mM N peptide-boxB RNA complex; 25mM D6(98%)-succinate, 2mM NaCl, 0.2mM EDTA, 0.05mM Na-azide Solvent system: 90% H20, 10% D2O |
|---|---|
| Sample conditions | Ionic strength: 2mM NaCl / pH: 6.0 / Pressure: ambient / Temperature: 298 K |
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Radiation wavelength | Relative weight: 1 | ||||||||||||||||||||
| NMR spectrometer |
|
- Processing
Processing
| NMR software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Software ordinal: 1 Details: There were a total of 1016 distance restraints (including 48 hydrogen bond distance restraints) and 167 torsion restraints used to determine this structure. The molecular modeling of the ...Details: There were a total of 1016 distance restraints (including 48 hydrogen bond distance restraints) and 167 torsion restraints used to determine this structure. The molecular modeling of the phi21 N peptide-boxB RNA complex was done in three steps. First, a complete intramolecular restraint set was generated for each molecule, and then peptide and RNA structures were generated separately using ab initio simulated annealing (SA) starting from a random extended structure in CNSsolve. For both the peptide and the RNA, constrained (torsion) dynamics was used at 50000K. A total of 100 structures each of the peptide and RNA were generated. In the second step, each of the 20 lowest energy peptide and 20 lowest energy RNA structures were combined in single PDB files, in all 400 possible combinations. The RNA was held at the origin and the peptide was randomly rotated and moved 100 angstroms away in a random direction from the origin. These 400 possible "complexes" were docked using CNSsolve. The objective of the docking was to bring the peptide and RNA together without dramatically perturbing their folded structures from the first round of SA, so the temperatures for the docking were set much lower than in the initial calculations (1000 vs. 50000K). Finally, the 100 lowest energy docked structures were minimized by two rounds of low temperature annealing using Sander, a module of AMBER. As with the docking, the temperature was kept low (1000K). The 14 lowest energy structures were used for generating an average structure, which was energy minimized using a conjugate gradient. | ||||||||||||||||||||
| NMR representative | Selection criteria: minimized average structure | ||||||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 100 / Conformers submitted total number: 15 |
