 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nnk: X-ray structure of the GluR2 ligand-binding core (S1S2J) in compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nnk | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of the GluR2 ligand-binding core (S1S2J) in complex with (S)-ATPA at 1.85 A resolution. Crystallization with zinc ions. | ||||||
 Components Components | Glutamate receptor 2 GRIA2 GRIA2 | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / Ionotropic glutamate receptor GluR2 / ligand-binding core / agonist complex | ||||||
| Function / homology |  Function and homology information Function and homology informationspine synapse / dendritic spine neck / dendritic spine head / Activation of AMPA receptors / response to lithium ion / perisynaptic space / cellular response to glycine / AMPA glutamate receptor activity / Trafficking of GluR2-containing AMPA receptors / immunoglobulin binding ...spine synapse / dendritic spine neck / dendritic spine head / Activation of AMPA receptors / response to lithium ion / perisynaptic space / cellular response to glycine / AMPA glutamate receptor activity / Trafficking of GluR2-containing AMPA receptors / immunoglobulin binding / AMPA glutamate receptor complex / kainate selective glutamate receptor activity / ionotropic glutamate receptor complex / extracellularly glutamate-gated ion channel activity / asymmetric synapse / regulation of receptor recycling / Unblocking of NMDA receptors, glutamate binding and activation / glutamate receptor binding / positive regulation of synaptic transmission / glutamate-gated receptor activity / presynaptic active zone membrane / response to fungicide / regulation of synaptic transmission, glutamatergic / cellular response to brain-derived neurotrophic factor stimulus / somatodendritic compartment / dendrite membrane / ligand-gated monoatomic ion channel activity involved in regulation of presynaptic membrane potential / ionotropic glutamate receptor binding / ionotropic glutamate receptor signaling pathway / dendrite cytoplasm / cytoskeletal protein binding / SNARE binding / transmitter-gated monoatomic ion channel activity involved in regulation of postsynaptic membrane potential / dendritic shaft / synaptic membrane / synaptic transmission, glutamatergic / PDZ domain binding / postsynaptic density membrane / protein tetramerization / modulation of chemical synaptic transmission / Schaffer collateral - CA1 synapse / establishment of protein localization / terminal bouton / receptor internalization / synaptic vesicle membrane / cerebral cortex development / synaptic vesicle / presynapse / signaling receptor activity / presynaptic membrane / amyloid-beta binding / growth cone / perikaryon / chemical synaptic transmission / scaffold protein binding / postsynaptic membrane / dendritic spine / postsynaptic density / neuron projection / axon / neuronal cell body / dendrite / synapse / glutamatergic synapse / protein-containing complex binding / endoplasmic reticulum membrane / protein kinase binding / cell surface / endoplasmic reticulum / protein-containing complex / membrane / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Rattus norvegicus (Norway rat) Rattus norvegicus (Norway rat) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Lunn, M.-L. / Hogner, A. / Stensbol, T.B. / Gouaux, E. / Egebjerg, J. / Kastrup, J.S. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2003 Title: Three-Dimensional Structure of the Ligand-Binding Core of GluR2 in Complex with the Agonist (S)-ATPA: Implications for Receptor Subunit Selectivity. Authors: Lunn, M.L. / Hogner, A. / Stensbol, T.B. / Gouaux, E. / Egebjerg, J. / Kastrup, J.S. #1: Journal: J.Mol.Biol. / Year: 2002Title: Structural basis for AMPA receptor activation and ligand selectivity: Crystal structures of five agonist complexes with the GluR2 ligand binding core. Authors: Hogner, A. / Kastrup, J.S. / Jin, R. / Liljefors, T. / Mayer, M.L. / Egebjerg, J. / Larsen, I. / Gouaux, E. #2: Journal: Neuron / Year: 2000Title: Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Authors: Armstrong, N. / Gouaux, E. #3: Journal: Nature / Year: 2002Title: Mechanism of glutamate receptor desensitization. Authors: Sun, Y. / Olson, R. / Horning, M. / Armstrong, N. / Mayer, M. / Gouaux, E. #4: Journal: Protein Sci. / Year: 1998Title: Probing the ligand binding domain of the GluR2 receptor by proteolysis and deletion mutagenesis defines domain boundaries and yields a crystallizable construct. Authors: Chen, G.Q. / Sun, R. / Jin, R. / Gouaux, E. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 ... BIOMOLECULE: 1 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 1 CHAIN(S). SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). NOTE THAT COORDINATES FOR ONE DIMER OF THE TETRAMERIC MULTIMER REPRESENTING THE KNOWN BIOLOGICALLY SIGNIFICANT OLIGOMERIZATION STATE OF THE MOLECULE CAN BE GENERATED BY APPLYING BIOMT TRANSFORMATIONS GIVEN IN REMARK 350. | ||||||
| Remark 999 | SEQUENCE Native GluR2 is a membrane protein. The protein crystallized is the extracellular ligand- ...SEQUENCE Native GluR2 is a membrane protein. The protein crystallized is the extracellular ligand-binding core of GluR2. Transmembrane regions were genetically removed and replaced with a Gly-Thr linker (residues 115-116). Therefore, the sequence matches discontinuously with the reference database (413-527, 653-796). The two first residues of the sequence (Gly-2, Ala-1) are cloning artifacts and were not located in the electron density map. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nnk.cif.gz | 72.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nnk.ent.gz | 52.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1nnk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nn/1nnkftp://data.pdbj.org/pub/pdb/validation_reports/nn/1nnk | HTTPS FTP |
|---|
-Related structure data









































-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer composed of dimers-of-dimers. Only the dimer is observed in the crystal. The dimer may be generated by applying the following to chain A: TRANSFORM FRACTIONAL - -1.00000 0.00000 0.00000 - 0.00000 -1.00000 0.00000 - 0.00000 0.00000 1.00000 - 2.00000 0.00000 0.00000 |
-Components
| #1: Protein | GRIA2 / GLUR-2 / GLUR-B / GLUTAMATE RECEPTOR IONOTROPIC AMPA 2 Mass: 29221.682 Da / Num. of mol.: 1 / Fragment: GluR2-flop ligand-binding core (S1S2J) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rattus norvegicus (Norway rat) / Plasmid: pET30B / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P19491 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P19491 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-CL / | Chloride  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-CE2 / | 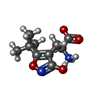  Mass: 227.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O4 Mass: 227.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O4#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 179 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 49 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 279 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: zinc acetate, cacodylate, PEG8000, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 279K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 6 ℃ | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I711 / Wavelength: 1.0835 Å / Beamline: I711 / Wavelength: 1.0835 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 14, 2001 |
| Radiation | Monochromator: NULL. / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0835 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→20 Å / Num. all: 23016 / Num. obs: 23016 / % possible obs: 92.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.5 % / Biso Wilson estimate: 18.2 Å2 / Rmerge(I) obs: 0.099 / Net I/σ(I): 7 |
| Reflection shell | Resolution: 1.85→1.92 Å / Rmerge(I) obs: 0.34 / Mean I/σ(I) obs: 2 / Num. unique all: 1996 / % possible all: 82.1 |
| Reflection | *PLUS Lowest resolution: 20 Å |
| Reflection shell | *PLUS % possible obs: 82.1 % / Rmerge(I) obs: 0.34 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: GluR2:(S)-thio-ATPA complex (Lunn et al., to be published). Resolution: 1.85→19.99 Å / Rfactor Rfree error: 0.009 / Data cutoff high rms absF: 1449707.81 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: The first three N-terminal residues and the last two C-terminal residues were not located in the electron density map.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 48.7733 Å2 / ksol: 0.418009 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.4 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→19.99 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.85→1.97 Å / Rfactor Rfree error: 0.029 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: protein_rep.param / Topol file: protein.top | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / % reflection Rfree: 2.7 % | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
