 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
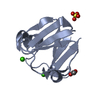
Yorodumi- PDB-1kyc: CRYSTAL STRUCTURE OF A DE NOVO DESIGNED TRIMERIC COILED-COIL PEPT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kyc | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A DE NOVO DESIGNED TRIMERIC COILED-COIL PEPTIDE STABLIZED BY IONIC INTERACTIONS | ||||||
 Components Components | SIN-GLU-GLU-LEU-ARG-ARG-ARG-ILE-GLU-GLU-LEU-GLU-ARG-ARG-ILE-ARG-NH2 | ||||||
 Keywords Keywords |  DE NOVO PROTEIN / COILED COIL / DE NOVO DESIGN / ALPHA-HELIX / TRIMER DE NOVO PROTEIN / COILED COIL / DE NOVO DESIGN / ALPHA-HELIX / TRIMER | ||||||
| Function / homology | SUCCINIC ACID Function and homology information Function and homology information | ||||||
| Biological species | synthetic construct (others) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Burkhard, P. / Ivaninskii, S. / Lustig, A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Improving coiled-coil stability by optimizing ionic interactions. Authors: Burkhard, P. / Ivaninskii, S. / Lustig, A. #1: Journal: Protein Sci. / Year: 2000Title: Design of a Minimal Protein Oligomerization Domain by a Structural Approach Authors: Burkhard, P. / Meier, M. / Lustig, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kyc.cif.gz | 17.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kyc.ent.gz | 11.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1kyc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ky/1kycftp://data.pdbj.org/pub/pdb/validation_reports/ky/1kyc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hqjS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein/peptide | Mass: 2057.386 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: This peptide was chemically synthesized. / Source: (synth.) synthetic construct (others) | ||||
|---|---|---|---|---|---|
| #2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | Succinic acid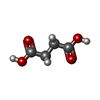  Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.71 Å3/Da / Density % sol: 28.17 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: ammonium sulfate, HEPES, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ELLIOTT GX-21 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 5, 2001 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→30 Å / Num. obs: 2574 / % possible obs: 96.2 % / Observed criterion σ(I): -3 / Redundancy: 10 % / Rsym value: 0.54 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 1.45→1.5 Å / Redundancy: 2.3 % / Mean I/σ(I) obs: 7 / Rsym value: 0.131 / % possible all: 72.1 |
| Reflection | *PLUS Highest resolution: 1.45 Å / Lowest resolution: 30 Å / % possible obs: 96.9 % / Num. measured all: 47109 / Rmerge(I) obs: 0.54 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HQJ Resolution: 1.45→30 Å / Num. parameters: 1680 / Num. restraintsaints: 2133 / σ(F): 0
| ||||||||||||||||
| Refine analyze | Num. disordered residues: 1 / Occupancy sum hydrogen: 1384 / Occupancy sum non hydrogen: 1815 | ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→30 Å
| ||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.217 / Rfactor Rwork: 0.166 | ||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||
| Refine LS restraints | *PLUS
|
