 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dxm | ||||||
|---|---|---|---|---|---|---|---|
| Title | Reduced form of the H protein from glycine decarboxylase complex | ||||||
 Components Components | H PROTEIN | ||||||
 Keywords Keywords | OXIDOREDUCTASES(ACTING ON CH-NH2 DONOR) /  GLYCINE DECARBOXYLASE / MITOCHONDRIA GLYCINE DECARBOXYLASE / MITOCHONDRIA | ||||||
| Function / homology |  Function and homology informationglycine cleavage complex / glycine decarboxylation via glycine cleavage system / mitochondrion Function and homology informationglycine cleavage complex / glycine decarboxylation via glycine cleavage system / mitochondrionSimilarity search - Function | ||||||
| Biological species |   PISUM SATIVUM (garden pea) PISUM SATIVUM (garden pea) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Faure, M. / Cohen-Addad, C. / Neuburger, M. / Douce, R. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 2000 Title: Interaction between the Lipoamide-Containing H-Protein and the Lipoamide Dehydrogenase (L-Protein) of the Glycine Decarboxylase Multienzyme System. 2. Crystal Structure of H- and L-Proteins Authors: Faure, M. / Bourguignon, J. / Neuburger, M. / Macherel, D. / Sieker, L. / Ober, R. / Kahn, R. / Cohen-Addad, C. / Douce, R. #1: Journal: Biochimie / Year: 1997 Title: Structural Studies of the Glycine Decarboxylase Complex from Pea Leaf Mitochondria Authors: Cohen-Addad, C. / Faure, M. / Neuburger, M. / Ober, R. / Sieker, L. / Bourguignon, J. / Macherel, D. / Douce, R. #2: Journal: Nat.Struct.Biol. / Year: 1995Title: The Lipoamide Arm in the Glycine Decarboxylase is not Freely Swinging Authors: Cohen-Addad, C. / Pares, S. / Sieker, L. / Neuburger, M. / Douce, R. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1995Title: Refined Structures at 2 and 2.2 A Resolution of Two Forms of the H-Protein, a Lipoamide-Containing Protein of the Glycine Decarboxylase Complex Authors: Pares, S. / Cohen-Addad, C. / Sieker, L. / Neuburger, M. / Douce, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dxm.cif.gz | 64.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dxm.ent.gz | 48.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1dxm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dx/1dxmftp://data.pdbj.org/pub/pdb/validation_reports/dx/1dxm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1dxlC  1hpcS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.39443, 0.54761, 0.73794), Vector : Details | BIOLOGICAL_UNIT: MONOMERTHE GLYCINE CLEAVAGE SYSTEM IS COMPOSED OF FOUR PROTEINS:P, T, L, AND H. THE H CHAIN WAS STUDIED HERE. THIS COMPONENTSHUTTLES THE METHYLAMINE GROUP OF GLYCINE FROM THE P PROTEINTO THE T PROTEIN. THE H CHAIN CONTAINS A COVALENTLY-BOUNDLIPOYL COFACTOR. | |
-Components
| #1: Protein | Mass: 13962.464 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PISUM SATIVUM (garden pea) / Organ: LEAF / Organelle: MITOCHONDRIAMitochondrion / Plasmid: PET-HM / Cellular location (production host): CYTOPLASM / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P16048 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P16048#2: Chemical | Dihydrolipoic acid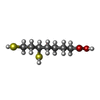  Mass: 208.341 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H16O2S2 Mass: 208.341 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H16O2S2#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 145 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 145 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.67 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.2 Details: 50 % AMMONIUM SULFATE, 100 MM TRIS MALEATE PH 5.2, 2 MM TCEP (TRIS CARBOXYETHYL PHOSPHINE) | ||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 8 ℃ / Method: vapor diffusion | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Wavelength: 1.5418 |
| Detector | Detector: IMAGE PLATE / Date: Dec 15, 1998 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→40 Å / Num. obs: 7808 / % possible obs: 97.1 % / Observed criterion σ(I): 0 / Redundancy: 3 % / Biso Wilson estimate: 47.6 Å2 / Rsym value: 0.11 / Net I/σ(I): 4.7 |
| Reflection shell | Resolution: 2.6→2.74 Å / Redundancy: 2.9 % / Mean I/σ(I) obs: 3 / Rsym value: 0.24 / % possible all: 98.3 |
| Reflection | *PLUS % possible obs: 97 % / Rmerge(I) obs: 0.11 |
| Reflection shell | *PLUS % possible obs: 98.3 % / Rmerge(I) obs: 0.24 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HPC Resolution: 2.6→40 Å / Rfactor Rfree error: 0.013 / Data cutoff high absF: 1374190.82 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 48 Å2 / ksol: 0.358993 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.76 Å / Rfactor Rfree error: 0.046 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARHCSDX.PRO / Topol file: TOPHCSDX.PRO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.207 / Rfactor Rfree: 0.261 / Rfactor Rwork: 0.207 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
