 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bu5: X-RAY CRYSTAL STRUCTURE OF THE DESULFOVIBRIO VULGARIS (HILDENBORO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bu5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-RAY CRYSTAL STRUCTURE OF THE DESULFOVIBRIO VULGARIS (HILDENBOROUGH) APOFLAVODOXIN-RIBOFLAVIN COMPLEX | ||||||
 Components Components | PROTEIN (FLAVODOXIN) | ||||||
 Keywords Keywords |  ELECTRON TRANSPORT / FLAVOPROTEIN ELECTRON TRANSPORT / FLAVOPROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Desulfovibrio vulgaris (bacteria) Desulfovibrio vulgaris (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.83 Å | ||||||
 Authors Authors | Walsh, M.A. / Mccarthy, A. / O'Farrell, P.A. / Mccardle, P. / Cunningham, P.D. / Mayhew, S.G. / Higgins, T.M. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1998 Title: X-ray crystal structure of the Desulfovibrio vulgaris (Hildenborough) apoflavodoxin-riboflavin complex. Authors: Walsh, M.A. / McCarthy, A. / O'Farrell, P.A. / McArdle, P. / Cunningham, P.D. / Mayhew, S.G. / Higgins, T.M. #1: Journal: J.Mol.Biol. / Year: 1991Title: Comparison of the Crystal Structures of a Flavodoxin in its Three Oxidation States at Cryogenictemperatures Authors: Watt, W. / Tulinsky, A. / Swenson, R.P. / Watenpaugh, K.D. #2: Journal: J.Biol.Chem. / Year: 1988Title: Cloning, Nucleotide Sequence, and Expression of the Flavodoxin Gene from Desulfovibrio Vulgaris (Hildenborough) Authors: Krey, G.D. / Vanin, E.F. / Swenson, R.P. #3: Journal: Fems Microbiol.Lett. / Year: 1988Title: Cloning and Sequencing of the Gene Encoding Flavodoxin from Desulfovibrio Vulgaris Hildenborough Authors: Curley, G.P. / Voordouw, G. #4: Journal: Proc.Natl.Acad.Sci.USA / Year: 1973Title: The Binding of Riboflavin-5-Phosphate in a Flavoprotein. Flavodoxin at 2.0- Angstroms Resolution Authors: Watenpaugh, K.D. / Sieker, L.C. / Jensen, L.H. #5: Journal: Proc.Natl.Acad.Sci.USA / Year: 1972Title: Structure of the Oxidized Form of a Flavodoxin at 2.5-Angstroms Resolution. Resolution of the Phase Ambiguity by Anomalous Scattering Authors: Watenpaugh, K.D. / Sieker, L.C. / Jensen, L.H. / Legall, J. / Dubourdieu, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bu5.cif.gz | 74.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bu5.ent.gz | 55.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1bu5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bu/1bu5ftp://data.pdbj.org/pub/pdb/validation_reports/bu/1bu5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2fx2S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.38919, -0.92107, 0.01285), Vector : |
-Components
| #1: Protein | Mass: 15704.146 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Desulfovibrio vulgaris (bacteria) / Production host: Escherichia coli (E. coli) / Strain (production host): TG1 / References: UniProt: P00323#2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | Riboflavin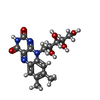  Mass: 376.364 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H20N4O6 Mass: 376.364 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H20N4O6#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.75 Å3/Da / Density % sol: 55 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: CRYSTALS WERE GROWN USING THE HANGING DROP METHOD OF VAPOUR DIFFUSION FROM TRIALS WITH AMMONIUM SULPHATE IN THE CONCENTRATION RANGE 60-80% SATURATION IN 50MM SODIUM PHOSPHATE BUFFER PH 8.0 ...Details: CRYSTALS WERE GROWN USING THE HANGING DROP METHOD OF VAPOUR DIFFUSION FROM TRIALS WITH AMMONIUM SULPHATE IN THE CONCENTRATION RANGE 60-80% SATURATION IN 50MM SODIUM PHOSPHATE BUFFER PH 8.0 CONTAINING 1MM EDTA WITH A PROTEIN CONCENTRATION OF 10-15MG/ML. CRYSTALS APPEAR OVER 1-3 DAYS., pH 7.0 | |||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 55 % | |||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.5 / Wavelength: 0.9 / Beamline: PX9.5 / Wavelength: 0.9 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1.83→20 Å / Num. obs: 26539 / % possible obs: 79.9 % / Observed criterion σ(I): -3 / Redundancy: 1.5 % / Biso Wilson estimate: 23 Å2 / Rmerge(I) obs: 0.042 / Net I/σ(I): 15.3 |
| Reflection shell | Resolution: 1.83→1.86 Å / Redundancy: 1.5 % / Rmerge(I) obs: 0.23 / Mean I/σ(I) obs: 2.5 / % possible all: 83.2 |
| Reflection | *PLUS Num. measured all: 72050 |
| Reflection shell | *PLUS % possible obs: 83.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: WILD TYPE FLAVODOXIN COORDINATES NOT DEPOSITED [M.A.WALSH PH.D THESIS NATIONAL UNIVERSITY OF IRELAND (1994)] BUT ESSENTIALLY IDENTICAL TO PDB ENTRY 2FX2 Resolution: 1.83→10 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 10 Å / Luzzati sigma a obs: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.83→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CCP4 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 0.08 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 27.1 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
