 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1b59: COMPLEX OF HUMAN METHIONINE AMINOPEPTIDASE-2 COMPLEXED WITH OVALICIN -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b59 | ||||||
|---|---|---|---|---|---|---|---|
| Title | COMPLEX OF HUMAN METHIONINE AMINOPEPTIDASE-2 COMPLEXED WITH OVALICIN | ||||||
 Components Components | PROTEIN (METHIONINE AMINOPEPTIDASE) | ||||||
 Keywords Keywords |  HYDROLASE / ANGIOGENESIS INHIBITOR HYDROLASE / ANGIOGENESIS INHIBITOR | ||||||
| Function / homology |  Function and homology informationN-terminal protein amino acid modification / peptidyl-methionine modification / initiator methionyl aminopeptidase activity / methionyl aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / protein processing / Inactivation, recovery and regulation of the phototransduction cascade / RNA binding ...N-terminal protein amino acid modification / peptidyl-methionine modification / initiator methionyl aminopeptidase activity / methionyl aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / protein processing / Inactivation, recovery and regulation of the phototransduction cascade / RNA binding / metal ion binding / plasma membrane / cytosol / cytoplasm Function and homology informationN-terminal protein amino acid modification / peptidyl-methionine modification / initiator methionyl aminopeptidase activity / methionyl aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / protein processing / Inactivation, recovery and regulation of the phototransduction cascade / RNA binding ...N-terminal protein amino acid modification / peptidyl-methionine modification / initiator methionyl aminopeptidase activity / methionyl aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / protein processing / Inactivation, recovery and regulation of the phototransduction cascade / RNA binding / metal ion binding / plasma membrane / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Liu, S. / Clardy, J.C. | ||||||
 Citation Citation | Journal: Science / Year: 1998 Title: Structure of human methionine aminopeptidase-2 complexed with fumagillin. Authors: Liu, S. / Widom, J. / Kemp, C.W. / Crews, C.M. / Clardy, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b59.cif.gz | 89.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b59.ent.gz | 65.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1b59.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b5/1b59ftp://data.pdbj.org/pub/pdb/validation_reports/b5/1b59 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b6aC  1bn5SC  1boaC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41453.090 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: OVALICIN COVALENTLY LINKED TO HIS 231 NE2 / Source: (gene. exp.) Homo sapiens (human)Description: PROTEIN WAS EXPRESSED IN SF21 INSECT CELLS.. BACULOVIRUS PACSG2 VECTOR Cellular location: CYTOPLASM / Genus (production host): Nucleopolyhedrovirus / Cell line (production host): SF21 / Production host:  Spodoptera frugiperda MNPV (virus) / Strain (production host): SF21 / References: UniProt: P50579, methionyl aminopeptidase Spodoptera frugiperda MNPV (virus) / Strain (production host): SF21 / References: UniProt: P50579, methionyl aminopeptidase | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co#3: Chemical | ChemComp-OVA / | 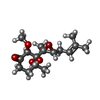  Mass: 298.375 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H26O5 Mass: 298.375 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H26O5#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 213 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.12 Å3/Da / Density % sol: 42 % |
|---|---|
| Crystal grow | pH: 5.4 / Details: 15-30% T-BUTANOL, 50MM CITRATE BUFFER PH=5.2-5.4 |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F2 / Wavelength: 0.979 / Beamline: F2 / Wavelength: 0.979 |
| Detector | Type: ADSC / Detector: CCD / Date: Aug 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→25 Å / Num. obs: 40120 / % possible obs: 96.3 % / Redundancy: 5.1 % / Biso Wilson estimate: 22.7 Å2 / Rmerge(I) obs: 0.074 / Rsym value: 0.074 / Net I/σ(I): 15.2 |
| Reflection shell | Resolution: 1.8→1.9 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.298 / Mean I/σ(I) obs: 3.9 / Rsym value: 0.298 / % possible all: 97.7 |
| Reflection | *PLUS Lowest resolution: 25 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BN5 Resolution: 1.8→6 Å / Rfactor Rfree error: 0.005 / Data cutoff high rms absF: 2190766.89 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / ksol: 0.762 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.91 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.4 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5.1 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 27.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.303 / % reflection Rfree: 5.1 % / Rfactor Rwork: 0.234 |
