 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-27903: Design of Diverse Asymmetric Pockets in de novo Homo-oligomeric P... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Design of Diverse Asymmetric Pockets in de novo Homo-oligomeric Proteins | |||||||||
 Map data Map data | Sharpened Map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Escherichia coli (E. coli) / synthetic construct (others) Escherichia coli (E. coli) / synthetic construct (others) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.85 Å | |||||||||
 Authors Authors | Gerben S / Borst AJ / Baker D | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2023 Title: Design of Diverse Asymmetric Pockets in Homo-oligomeric Proteins. Authors: Stacey R Gerben / Andrew J Borst / Derrick R Hicks / Isabelle Moczygemba / David Feldman / Brian Coventry / Wei Yang / Asim K Bera / Marcos Miranda / Alex Kang / Hannah Nguyen / David Baker / Abstract: A challenge for design of protein-small-molecule recognition is that incorporation of cavities with size, shape, and composition suitable for specific recognition can considerably destabilize protein ...A challenge for design of protein-small-molecule recognition is that incorporation of cavities with size, shape, and composition suitable for specific recognition can considerably destabilize protein monomers. This challenge can be overcome through binding pockets formed at homo-oligomeric interfaces between folded monomers. Interfaces surrounding the central homo-oligomer symmetry axes necessarily have the same symmetry and so may not be well suited to binding asymmetric molecules. To enable general recognition of arbitrary asymmetric substrates and small molecules, we developed an approach to designing asymmetric interfaces at off-axis sites on homo-oligomers, analogous to those found in native homo-oligomeric proteins such as glutamine synthetase. We symmetrically dock curved helical repeat proteins such that they form pockets at the asymmetric interface of the oligomer with sizes ranging from several angstroms, appropriate for binding a single ion, to up to more than 20 Å across. Of the 133 proteins tested, 84 had soluble expression in , 47 had correct oligomeric states in solution, 35 had small-angle X-ray scattering (SAXS) data largely consistent with design models, and 8 had negative-stain electron microscopy (nsEM) 2D class averages showing the structures coming together as designed. Both an X-ray crystal structure and a cryogenic electron microscopy (cryoEM) structure are close to the computational design models. The nature of these proteins as homo-oligomers allows them to be readily built into higher-order structures such as nanocages, and the asymmetric pockets of these structures open rich possibilities for small-molecule binder design free from the constraints associated with monomer destabilization. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_27903.map.gz | 38.1 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-27903-v30.xmlemd-27903.xml | 17.1 KB 17.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_27903_fsc.xml | 7.3 KB | Display | FSC data file |
| Images | 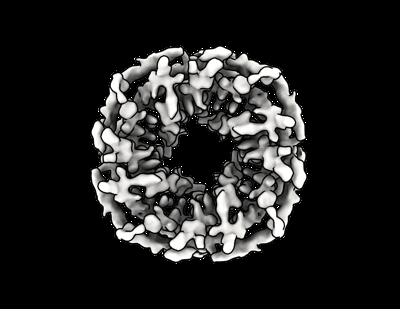 emd_27903.png emd_27903.png | 82.4 KB | ||
| Others | emd_27903_additional_1.map.gzemd_27903_half_map_1.map.gzemd_27903_half_map_2.map.gz | 19.4 MB 37.5 MB 37.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27903 http://ftp.pdbj.org/pub/emdb/structures/EMD-27903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27903 | HTTPS FTP |
-Related structure data


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_27903.map.gz / Format: CCP4 / Size: 40.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Sharpened Map | ||||||||||||||||||||
| Voxel size | X=Y=Z: 0.84 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
-Additional map: Unsharpened Map
| File | emd_27903_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened Map | ||||||||||||
| Projections & Slices |
| ||||||||||||
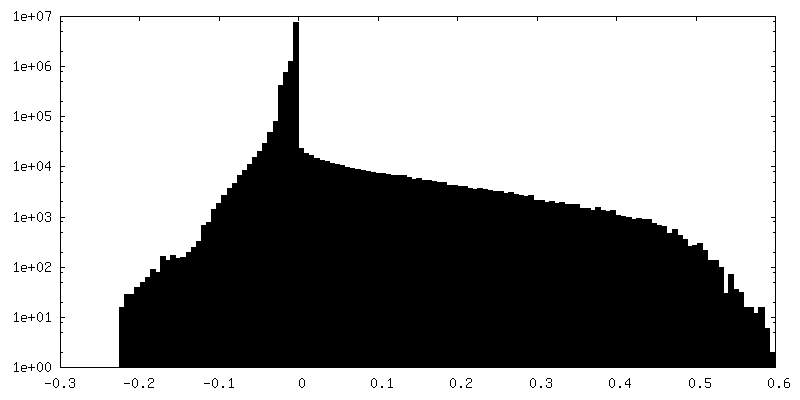
| Density Histograms |
 Z
Z Y
Y X
X







-Half map: Half Map A
| File | emd_27903_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half Map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





-Half map: Half Map B
| File | emd_27903_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half Map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : SG135
| Entire | Name: SG135 |
|---|---|
| Components |
|
-Supramolecule #1: SG135
| Supramolecule | Name: SG135 / type: complex / ID: 1 / Chimera: Yes / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) |
-Macromolecule #1: SG135
| Macromolecule | Name: SG135 / type: protein_or_peptide / ID: 1 Details: Deleted loop consisting of residues 173-179 due to lack of confident map density Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 23.521809 KDa |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: DREIKEEARK LIREAIELLQ KGDPRAKEIL RQAILILLAI RLLEEMEENI EKAEKLGNEE LSELAKRAIK LVREALELLK EGDPRAEEI LKLALKIIKA ILLLLEMYEN IKQAEELGDE DLSELAKIAI RLVRQALKLL QEGDPRAEEI LEIALRIIKL I LQLLFLKQ ...String: DREIKEEARK LIREAIELLQ KGDPRAKEIL RQAILILLAI RLLEEMEENI EKAEKLGNEE LSELAKRAIK LVREALELLK EGDPRAEEI LKLALKIIKA ILLLLEMYEN IKQAEELGDE DLSELAKIAI RLVRQALKLL QEGDPRAEEI LEIALRIIKL I LQLLFLKQ RIEEAKKKGD QQFVFEAEEK IRRIVEELFK LLEG |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.0 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: C-flat-1.2/1.3 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 1.7 µm / Nominal defocus min: 0.8 µm |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 63.56 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 2944810 |
|---|---|
| Startup model | Type of model: NONE / Details: Ab initio |
| Initial angle assignment | Type: NOT APPLICABLE / Software - Name: cryoSPARC (ver. 3.2) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC (ver. 3.2) |
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 3.85 Å / Resolution method: FSC 0.143 CUT-OFF Details: Removed large number over over-represented views from initial set of particles. Number images used: 855664 |
| FSC plot (resolution estimation) | 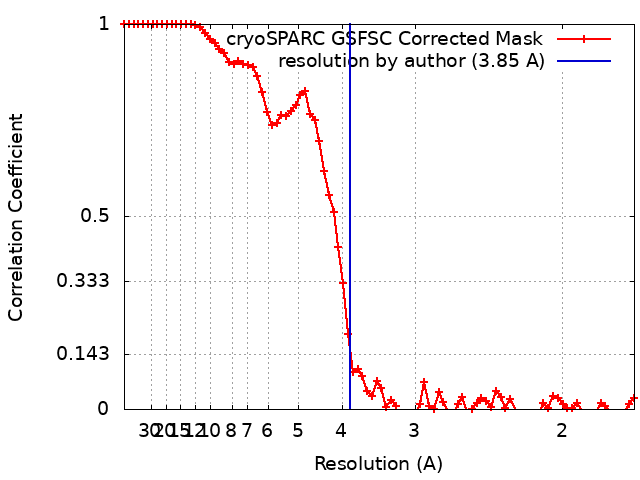 |
