 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Design of Diverse Asymmetric Pockets in de novo Homo-oligomeric Proteins | |||||||||
 マップデータ マップデータ | Sharpened Map | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |   Escherichia coli (大腸菌) / synthetic construct (人工物) Escherichia coli (大腸菌) / synthetic construct (人工物) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.85 Å | |||||||||
 データ登録者 データ登録者 | Gerben S / Borst AJ / Baker D | |||||||||
| 資金援助 |  米国, 1件 米国, 1件
| |||||||||
 引用 引用 | ジャーナル: Biochemistry / 年: 2023 タイトル: Design of Diverse Asymmetric Pockets in Homo-oligomeric Proteins. 著者: Stacey R Gerben / Andrew J Borst / Derrick R Hicks / Isabelle Moczygemba / David Feldman / Brian Coventry / Wei Yang / Asim K Bera / Marcos Miranda / Alex Kang / Hannah Nguyen / David Baker / 要旨: A challenge for design of protein-small-molecule recognition is that incorporation of cavities with size, shape, and composition suitable for specific recognition can considerably destabilize protein ...A challenge for design of protein-small-molecule recognition is that incorporation of cavities with size, shape, and composition suitable for specific recognition can considerably destabilize protein monomers. This challenge can be overcome through binding pockets formed at homo-oligomeric interfaces between folded monomers. Interfaces surrounding the central homo-oligomer symmetry axes necessarily have the same symmetry and so may not be well suited to binding asymmetric molecules. To enable general recognition of arbitrary asymmetric substrates and small molecules, we developed an approach to designing asymmetric interfaces at off-axis sites on homo-oligomers, analogous to those found in native homo-oligomeric proteins such as glutamine synthetase. We symmetrically dock curved helical repeat proteins such that they form pockets at the asymmetric interface of the oligomer with sizes ranging from several angstroms, appropriate for binding a single ion, to up to more than 20 Å across. Of the 133 proteins tested, 84 had soluble expression in , 47 had correct oligomeric states in solution, 35 had small-angle X-ray scattering (SAXS) data largely consistent with design models, and 8 had negative-stain electron microscopy (nsEM) 2D class averages showing the structures coming together as designed. Both an X-ray crystal structure and a cryogenic electron microscopy (cryoEM) structure are close to the computational design models. The nature of these proteins as homo-oligomers allows them to be readily built into higher-order structures such as nanocages, and the asymmetric pockets of these structures open rich possibilities for small-molecule binder design free from the constraints associated with monomer destabilization. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_27903.map.gz | 38.1 MB |  EMDBマップデータ形式 EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-27903-v30.xmlemd-27903.xml | 17.1 KB 17.1 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_27903_fsc.xml | 7.3 KB | 表示 | FSCデータファイル |
| 画像 | 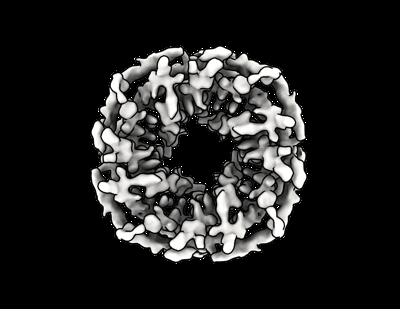 emd_27903.png emd_27903.png | 82.4 KB | ||
| その他 | emd_27903_additional_1.map.gzemd_27903_half_map_1.map.gzemd_27903_half_map_2.map.gz | 19.4 MB 37.5 MB 37.6 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27903 http://ftp.pdbj.org/pub/emdb/structures/EMD-27903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27903 | HTTPS FTP |
-関連構造データ


-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_27903.map.gz / 形式: CCP4 / 大きさ: 40.6 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Sharpened Map | ||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.84 Å | ||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||
| 詳細 | EMDB XML:
|
-添付データ
-追加マップ: Unsharpened Map
| ファイル | emd_27903_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened Map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
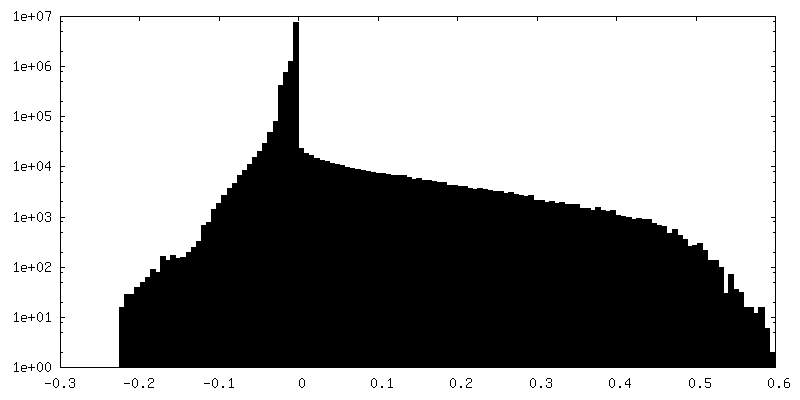
| 密度ヒストグラム |
 Z
Z Y
Y X
X







-ハーフマップ: Half Map A
| ファイル | emd_27903_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half Map A | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |





-ハーフマップ: Half Map B
| ファイル | emd_27903_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half Map B | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |






- 試料の構成要素
試料の構成要素
-全体 : SG135
| 全体 | 名称: SG135 |
|---|---|
| 要素 |
|
-超分子 #1: SG135
| 超分子 | 名称: SG135 / タイプ: complex / ID: 1 / キメラ: Yes / 親要素: 0 / 含まれる分子: all |
|---|---|
| 由来(天然) | 生物種: Escherichia coli (大腸菌) |
-分子 #1: SG135
| 分子 | 名称: SG135 / タイプ: protein_or_peptide / ID: 1 詳細: Deleted loop consisting of residues 173-179 due to lack of confident map density コピー数: 4 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: synthetic construct (人工物) |
| 分子量 | 理論値: 23.521809 KDa |
| 組換発現 | 生物種: Escherichia coli (大腸菌) |
| 配列 | 文字列: DREIKEEARK LIREAIELLQ KGDPRAKEIL RQAILILLAI RLLEEMEENI EKAEKLGNEE LSELAKRAIK LVREALELLK EGDPRAEEI LKLALKIIKA ILLLLEMYEN IKQAEELGDE DLSELAKIAI RLVRQALKLL QEGDPRAEEI LEIALRIIKL I LQLLFLKQ ...文字列: DREIKEEARK LIREAIELLQ KGDPRAKEIL RQAILILLAI RLLEEMEENI EKAEKLGNEE LSELAKRAIK LVREALELLK EGDPRAEEI LKLALKIIKA ILLLLEMYEN IKQAEELGDE DLSELAKIAI RLVRQALKLL QEGDPRAEEI LEIALRIIKL I LQLLFLKQ RIEEAKKKGD QQFVFEAEEK IRRIVEELFK LLEG |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1.0 mg/mL |
|---|---|
| 緩衝液 | pH: 7.5 |
| グリッド | モデル: C-flat-1.2/1.3 / 材質: COPPER / メッシュ: 400 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY |
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / 最大 デフォーカス(公称値): 1.7 µm / 最小 デフォーカス(公称値): 0.8 µm |
| 撮影 | フィルム・検出器のモデル: GATAN K3 (6k x 4k) / 平均電子線量: 63.56 e/Å2 |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 粒子像選択 | 選択した数: 2944810 |
|---|---|
| 初期モデル | モデルのタイプ: NONE / 詳細: Ab initio |
| 初期 角度割当 | タイプ: NOT APPLICABLE / ソフトウェア - 名称: cryoSPARC (ver. 3.2) |
| 最終 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: cryoSPARC (ver. 3.2) |
| 最終 再構成 | 解像度のタイプ: BY AUTHOR / 解像度: 3.85 Å / 解像度の算出法: FSC 0.143 CUT-OFF 詳細: Removed large number over over-represented views from initial set of particles. 使用した粒子像数: 855664 |
| FSC曲線 (解像度の算出) | 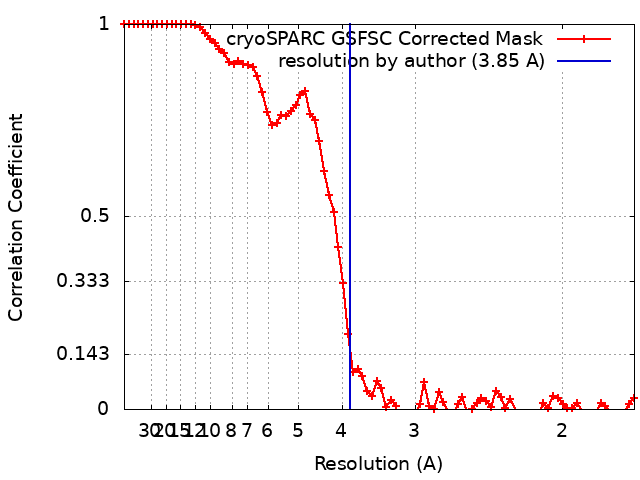 |
