 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-25836: CryoEM Structure of sFab COP-3 Complex with human claudin-4 and C... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM Structure of sFab COP-3 Complex with human claudin-4 and Clostridium perfringens enterotoxin C-terminal domain focused map | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  Claudin / cell adhesion / enterotoxin / Fab / antibody fragment / MEMBRANE PROTEIN Claudin / cell adhesion / enterotoxin / Fab / antibody fragment / MEMBRANE PROTEIN | |||||||||
| Function / homology | Clostridium enterotoxin / Clostridium enterotoxin / toxin activity / extracellular region / Heat-labile enterotoxin B chain Function and homology information Function and homology information | |||||||||
| Biological species |  Homo sapiens (human) / Homo sapiens (human) /   Clostridium perfringens (bacteria) Clostridium perfringens (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.8 Å | |||||||||
 Authors Authors | Vecchio AJ / Orlando BJ | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2022 Title: Development, structure, and mechanism of synthetic antibodies that target claudin and Clostridium perfringens enterotoxin complexes. Authors: Benjamin J Orlando / Pawel K Dominik / Sourav Roy / Chinemerem P Ogbu / Satchal K Erramilli / Anthony A Kossiakoff / Alex J Vecchio / Abstract: Strains of Clostridium perfringens produce a two-domain enterotoxin (CpE) that afflicts humans and domesticated animals, causing prevalent gastrointestinal illnesses. CpE's C-terminal domain (cCpE) ...Strains of Clostridium perfringens produce a two-domain enterotoxin (CpE) that afflicts humans and domesticated animals, causing prevalent gastrointestinal illnesses. CpE's C-terminal domain (cCpE) binds cell surface receptors, followed by a restructuring of its N-terminal domain to form a membrane-penetrating β-barrel pore, which is toxic to epithelial cells of the gut. The claudin family of membrane proteins are known receptors for CpE and also control the architecture and function of cell-cell contacts (tight junctions) that create barriers to intercellular molecular transport. CpE binding and assembly disables claudin barrier function and induces cytotoxicity via β-pore formation, disrupting gut homeostasis; however, a structural basis of this process and strategies to inhibit the claudin-CpE interactions that trigger it are both lacking. Here, we used a synthetic antigen-binding fragment (sFab) library to discover two sFabs that bind claudin-4 and cCpE complexes. We established these sFabs' mode of molecular recognition and binding properties and determined structures of each sFab bound to claudin-4-cCpE complexes using cryo-EM. The structures reveal that the sFabs bind a shared epitope, but conform distinctly, which explains their unique binding equilibria. Mutagenesis of antigen/sFab interfaces observed therein result in binding changes, validating the structures, and uncovering the sFab's targeting mechanism. From these insights, we generated a model for CpE's claudin-bound β-pore that predicted sFabs would not prevent cytotoxicity, which we then verified in vivo. Taken together, this work demonstrates the development and mechanism of claudin/cCpE-binding sFabs that provide a framework and strategy for obstructing claudin/CpE assembly to treat CpE-linked gastrointestinal diseases. | |||||||||
| History |
|
- Structure visualization
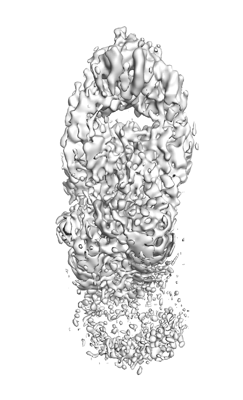
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_25836.map.gz | 115.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-25836-v30.xmlemd-25836.xml | 17 KB 17 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_25836_fsc.xml | 14.7 KB | Display | FSC data file |
| Images |  emd_25836.png emd_25836.png | 68.1 KB | ||
| Filedesc metadata | emd-25836.cif.gz | 5.1 KB | ||
| Others | emd_25836_half_map_1.map.gzemd_25836_half_map_2.map.gz | 116 MB 116 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25836ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25836 http://ftp.pdbj.org/pub/emdb/structures/EMD-25836ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25836 | HTTPS FTP |
-Related structure data




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_25836.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 0.871 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
-Half map: #1
| File | emd_25836_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X

-Half map: #2
| File | emd_25836_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
- Sample components
Sample components
-Entire : Human claudin-4/cCpE/COP-3 Complex
| Entire | Name: Human claudin-4/cCpE/COP-3 Complex |
|---|---|
| Components |
|
-Supramolecule #1: Human claudin-4/cCpE/COP-3 Complex
| Supramolecule | Name: Human claudin-4/cCpE/COP-3 Complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: COP-3 sFab fragment bound to Claudin-4/cCpE complex |
|---|---|
| Molecular weight | Theoretical: 85 kDa/nm |
-Macromolecule #1: COP-3 H chain
| Macromolecule | Name: COP-3 H chain / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: EISEVQLVES GGGLVQPGGS LRLSCAASGF NFYSSSIHWV RQAPGKGLEW VAYISSYSGY TYYADSVKGR FTISADTSKN TAYLQMNSL RAEDTAVYYC ARGYGYFDYN FSVGYALDYW GQGTLVTVSS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK D YFPEPVTV ...String: EISEVQLVES GGGLVQPGGS LRLSCAASGF NFYSSSIHWV RQAPGKGLEW VAYISSYSGY TYYADSVKGR FTISADTSKN TAYLQMNSL RAEDTAVYYC ARGYGYFDYN FSVGYALDYW GQGTLVTVSS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK D YFPEPVTV SWNSGALTSG VHTFPAVLQS SGLYSLSSVV TVPSSSLGTQ TYICNVNHKP SNTKVDKKVE PKSCDKTHT |
-Macromolecule #2: COP-3 L chain
| Macromolecule | Name: COP-3 L chain / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: SDIQMTQSPS SLSASVGDRV TITCRASQSV SSAVAWYQQK PGKAPKLLIY SASSLYSGVP SRFSGSRSGT DFTLTISSLQ PEDFATYYC QQSHPWYYPI TFGQGTKVEI KRTVAAPSVF IFPPSDSQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS G NSQESVTE ...String: SDIQMTQSPS SLSASVGDRV TITCRASQSV SSAVAWYQQK PGKAPKLLIY SASSLYSGVP SRFSGSRSGT DFTLTISSLQ PEDFATYYC QQSHPWYYPI TFGQGTKVEI KRTVAAPSVF IFPPSDSQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS G NSQESVTE QDSKDSTYSL SSTLTLSKAD YEKHKVYACE VTHQGLSSPV TKSFNRGEC |
-Macromolecule #3: Clostridium perfringens enterotoxin C-terminal domain
| Macromolecule | Name: Clostridium perfringens enterotoxin C-terminal domain / type: protein_or_peptide / ID: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Clostridium perfringens (bacteria) |
| Recombinant expression | Organism:  Trichoplusia ni (cabbage looper) Trichoplusia ni (cabbage looper) |
| Sequence | String: MSTDIEKEIL DLAAATERLN LTDALNSNPA GNLYDWRSSN SYPWTQKLNL HLTITATGQK YRILASKIVD FNIYSNNFNN LVKLEQSLGD GVKDHYVDIS LDAGQYVLVM KANSSYSGNY PYSILFQKFG LVPR UniProtKB: Heat-labile enterotoxin B chain |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 6.0 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Model: Quantifoil R1.2/1.3 / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 45 sec. / Pretreatment - Pressure: 0.001 kPa |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 2.2 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 120000 |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 3758631 / Average electron dose: 39.6 e/Å2 |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 305927 |
|---|---|
| Startup model | Type of model: PDB ENTRY PDB model - PDB ID: |
| Initial angle assignment | Type: OTHER |
| Final angle assignment | Type: OTHER |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.8 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: cryoSPARC / Number images used: 305927 |
| FSC plot (resolution estimation) | 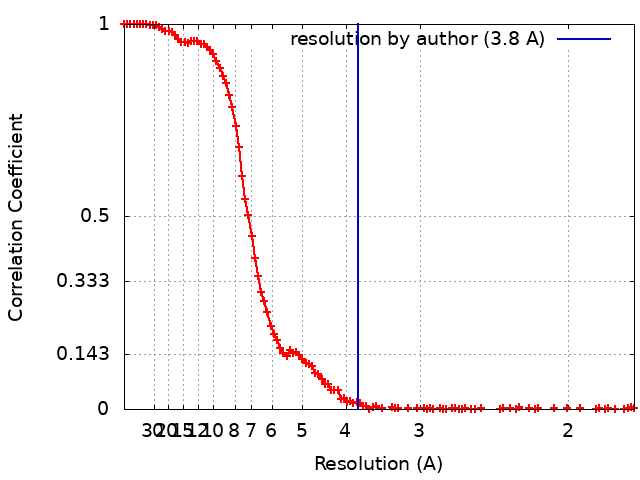 |
