 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-23355 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
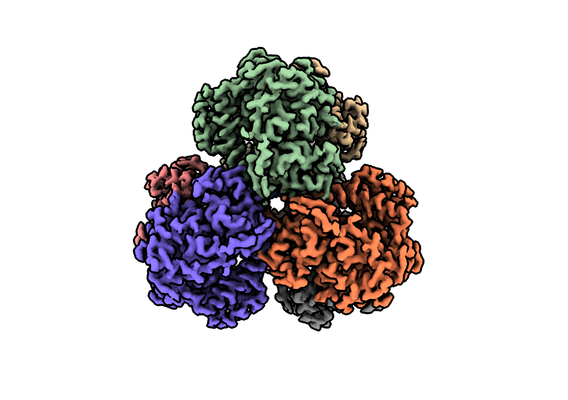
| タイトル | Structure of dNTPase at 3.3 Angstrom Resolution | |||||||||
 マップデータ マップデータ | Structure of dNTPase at 3.3 Angstrom Resolution | |||||||||
 試料 試料 |
| |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | |||||||||
| 生物種 |    Vibrio cholerae (コレラ菌) Vibrio cholerae (コレラ菌) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.3 Å | |||||||||
 データ登録者 データ登録者 | Bouvette J / Liu HF / Du X / Zhou Y / Sikkema AP / Mello JFR / Klemm B / Huang R / Schaaper RM / Borgnia MJ / Bartesaghi A | |||||||||
 引用 引用 | ジャーナル: Nat Commun / 年: 2021 タイトル: Beam image-shift accelerated data acquisition for near-atomic resolution single-particle cryo-electron tomography. 著者: Jonathan Bouvette / Hsuan-Fu Liu / Xiaochen Du / Ye Zhou / Andrew P Sikkema / Juliana da Fonseca Rezende E Mello / Bradley P Klemm / Rick Huang / Roel M Schaaper / Mario J Borgnia / Alberto Bartesaghi /  要旨: Tomographic reconstruction of cryopreserved specimens imaged in an electron microscope followed by extraction and averaging of sub-volumes has been successfully used to derive atomic models of ...Tomographic reconstruction of cryopreserved specimens imaged in an electron microscope followed by extraction and averaging of sub-volumes has been successfully used to derive atomic models of macromolecules in their biological environment. Eliminating biochemical isolation steps required by other techniques, this method opens up the cell to in-situ structural studies. However, the need to compensate for errors in targeting introduced during mechanical navigation of the specimen significantly slows down tomographic data collection thus limiting its practical value. Here, we introduce protocols for tilt-series acquisition and processing that accelerate data collection speed by up to an order of magnitude and improve map resolution compared to existing approaches. We achieve this by using beam-image shift to multiply the number of areas imaged at each stage position, by integrating geometrical constraints during imaging to achieve high precision targeting, and by performing per-tilt astigmatic CTF estimation and data-driven exposure weighting to improve final map resolution. We validated our beam image-shift electron cryo-tomography (BISECT) approach by determining the structure of a low molecular weight target (~300 kDa) at 3.6 Å resolution where density for individual side chains is clearly resolved. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_23355.map.gz | 3.6 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-23355-v30.xmlemd-23355.xml | 16.2 KB 16.2 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_23355_fsc.xml | 9.1 KB | 表示 | FSCデータファイル |
| 画像 | 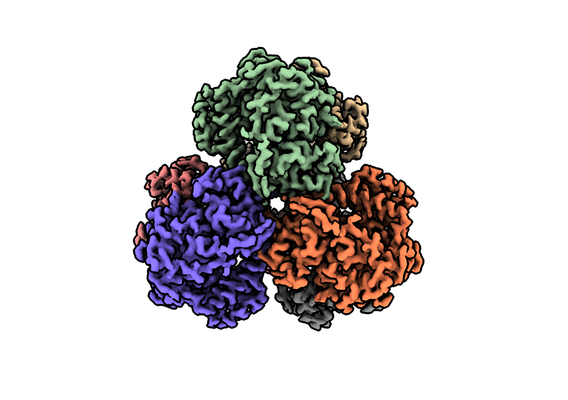 emd_23355.png emd_23355.png | 137.6 KB | ||
| マスクデータ | emd_23355_msk_1.map | 64 MB | マスクマップ | |
| その他 | emd_23355_additional_1.map.gzemd_23355_half_map_1.map.gzemd_23355_half_map_2.map.gz | 34.1 MB 34.1 MB 34.1 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23355ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23355 http://ftp.pdbj.org/pub/emdb/structures/EMD-23355ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23355 | HTTPS FTP |
-関連構造データ
| 関連構造データ | C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ |










-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_23355.map.gz / 形式: CCP4 / 大きさ: 3.9 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Structure of dNTPase at 3.3 Angstrom Resolution | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.37 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
-マスク #1
| ファイル | emd_23355_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
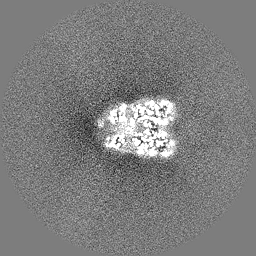
| 投影像・断面図 |
| ||||||||||||
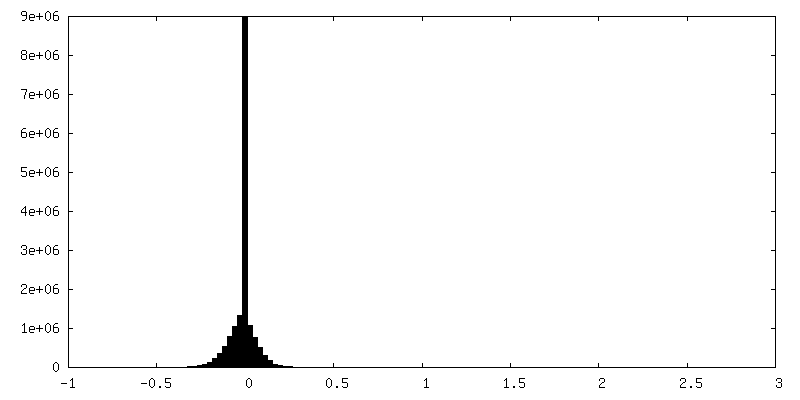
| 密度ヒストグラム |
 Z
Z Y
Y X
X







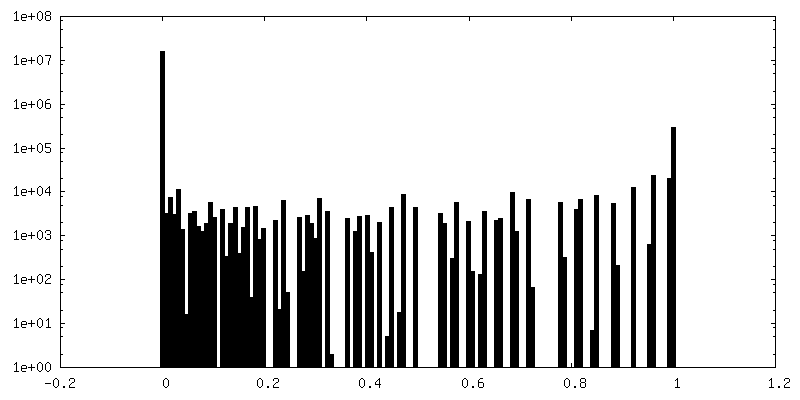
-追加マップ: Structure of dNTPase at 3.3 Angstrom Resolution
| ファイル | emd_23355_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Structure of dNTPase at 3.3 Angstrom Resolution | ||||||||||||
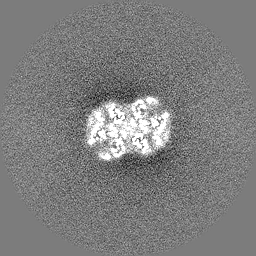
| 投影像・断面図 |
| ||||||||||||
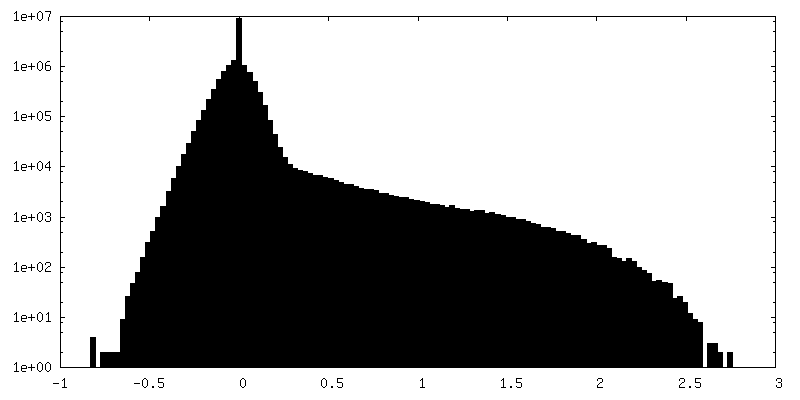
| 密度ヒストグラム |



-ハーフマップ: Structure of dNTPase at 3.3 Angstrom Resolution
| ファイル | emd_23355_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Structure of dNTPase at 3.3 Angstrom Resolution | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




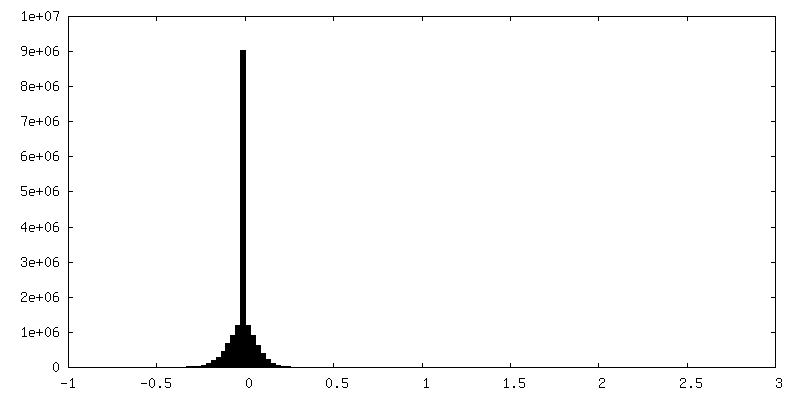
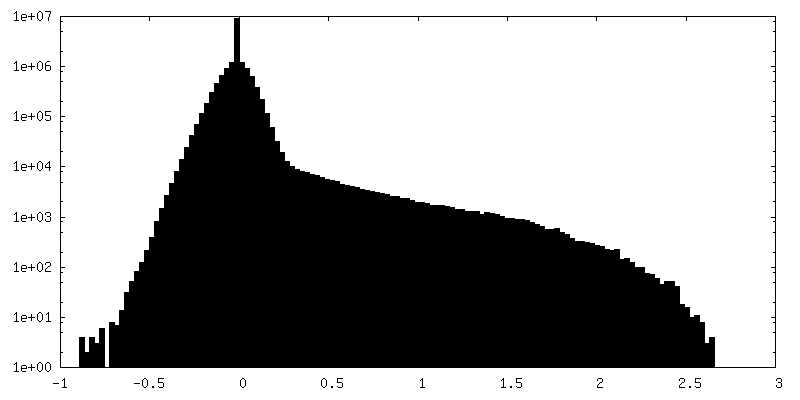
-ハーフマップ: Structure of dNTPase at 3.3 Angstrom Resolution
| ファイル | emd_23355_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Structure of dNTPase at 3.3 Angstrom Resolution | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |
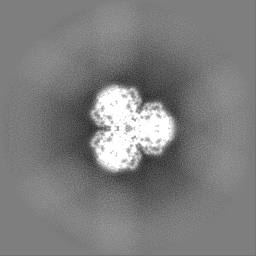
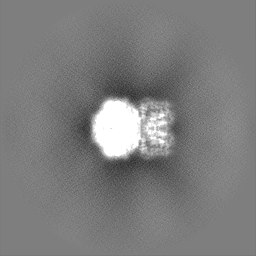


- 試料の構成要素
試料の構成要素
-全体 : dNTPase
| 全体 | 名称: dNTPase |
|---|---|
| 要素 |
|
-超分子 #1: dNTPase
| 超分子 | 名称: dNTPase / タイプ: complex / ID: 1 / 親要素: 0 |
|---|---|
| 由来(天然) | 生物種: Vibrio cholerae (コレラ菌) |
| 組換発現 | 生物種: Escherichia coli 'BL21-Gold(DE3)pLysS AG' (大腸菌) |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1.3 mg/mL |
|---|---|
| 緩衝液 | pH: 7.5 |
| グリッド | モデル: UltrAuFoil R1.2/1.3 / 材質: GOLD / メッシュ: 300 / 前処理 - タイプ: GLOW DISCHARGE |
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / 倍率(公称値): 105000 |
| 特殊光学系 | エネルギーフィルター - 名称: GIF Bioquantum / エネルギーフィルター - スリット幅: 20 eV |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / デジタル化 - サイズ - 横: 3838 pixel / デジタル化 - サイズ - 縦: 3710 pixel / デジタル化 - 画像ごとのフレーム数: 1-66 / 平均電子線量: 60.0 e/Å2 |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 粒子像選択 | 選択した数: 34190 |
|---|---|
| 初期 角度割当 | タイプ: PROJECTION MATCHING |
| 最終 角度割当 | タイプ: PROJECTION MATCHING |
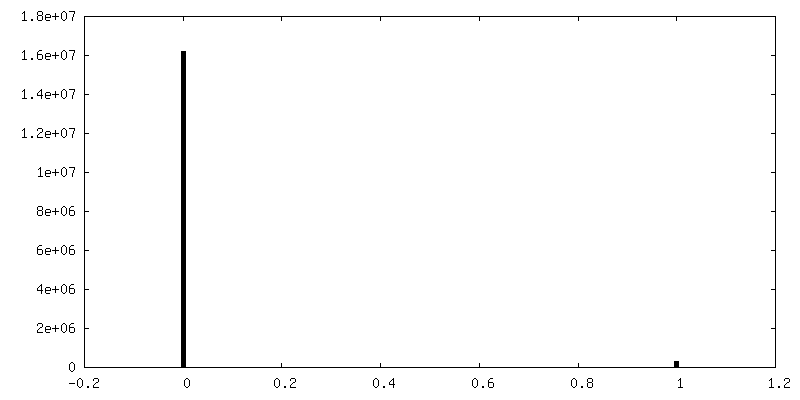
| 最終 再構成 | 使用したクラス数: 1 想定した対称性 - 点群: D3 (2回x3回 2面回転対称 )アルゴリズム: FOURIER SPACE / 解像度のタイプ: BY AUTHOR / 解像度: 3.3 Å / 解像度の算出法: FSC 0.143 CUT-OFF / 使用した粒子像数: 34190 |
| FSC曲線 (解像度の算出) |  |
