 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM structure of nucleotide-free GroEL-Rubisco. | |||||||||
 Map data Map data | CryoEM structure of GroEL bound to non-native Rubisco. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  GroEL / Rubisco / Chaperone / complex GroEL / Rubisco / Chaperone / complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat ...GroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat / protein refolding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosolSimilarity search - Function | |||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.4 Å | |||||||||
 Authors Authors | Gardner S / Saibil HR | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2023 Title: Structural basis of substrate progression through the bacterial chaperonin cycle. Authors: Scott Gardner / Michele C Darrow / Natalya Lukoyanova / Konstantinos Thalassinos / Helen R Saibil / Abstract: The bacterial chaperonin GroEL-GroES promotes protein folding through ATP-regulated cycles of substrate protein binding, encapsulation, and release. Here, we have used cryoEM to determine structures ...The bacterial chaperonin GroEL-GroES promotes protein folding through ATP-regulated cycles of substrate protein binding, encapsulation, and release. Here, we have used cryoEM to determine structures of GroEL, GroEL-ADP·BeF, and GroEL-ADP·AlF-GroES all complexed with the model substrate Rubisco. Our structures provide a series of snapshots that show how the conformation and interactions of non-native Rubisco change as it proceeds through the GroEL-GroES reaction cycle. We observe specific charged and hydrophobic GroEL residues forming strong initial contacts with non-native Rubisco. Binding of ATP or ADP·BeF to GroEL-Rubisco results in the formation of an intermediate GroEL complex displaying striking asymmetry in the ATP/ADP·BeF-bound ring. In this ring, four GroEL subunits bind Rubisco and the other three are in the GroES-accepting conformation, suggesting how GroEL can recruit GroES without releasing bound substrate. Our cryoEM structures of stalled GroEL-ADP·AlF-Rubisco-GroES complexes show Rubisco folding intermediates interacting with GroEL-GroES via different sets of residues. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_15939.map.gz | 120.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-15939-v30.xmlemd-15939.xml | 16.2 KB 16.2 KB | Display Display | EMDB header |
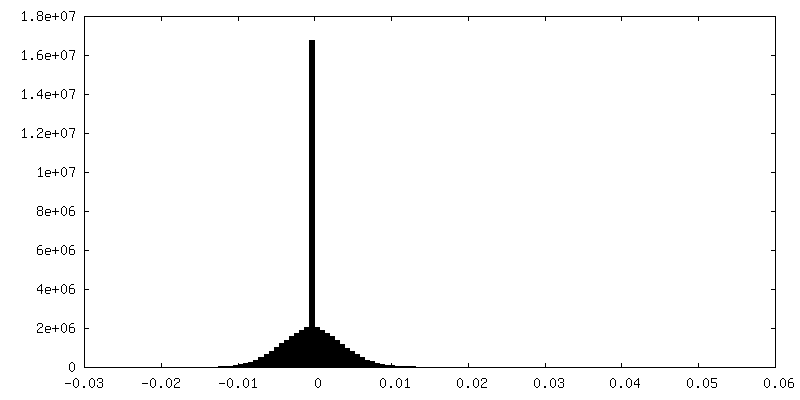
| FSC (resolution estimation) | emd_15939_fsc.xml | 13.6 KB | Display | FSC data file |
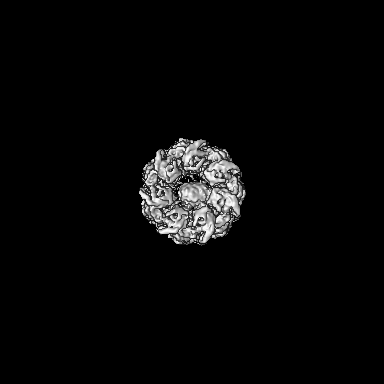
| Images |  emd_15939.png emd_15939.png | 108.6 KB | ||
| Masks | emd_15939_msk_1.map | 216 MB | Mask map | |
| Filedesc metadata | emd-15939.cif.gz | 5.7 KB | ||
| Others | emd_15939_half_map_1.map.gzemd_15939_half_map_2.map.gz | 172.5 MB 172.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-15939ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15939 http://ftp.pdbj.org/pub/emdb/structures/EMD-15939ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15939 | HTTPS FTP |
-Related structure data
| Related structure data |  8ba7MC  8ba8C  8ba9C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_15939.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM structure of GroEL bound to non-native Rubisco. | ||||||||||||||||||||||||||||||||||||
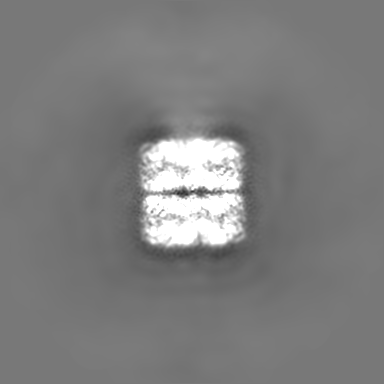
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.34 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)











-Supplemental data
-Mask #1
| File | emd_15939_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
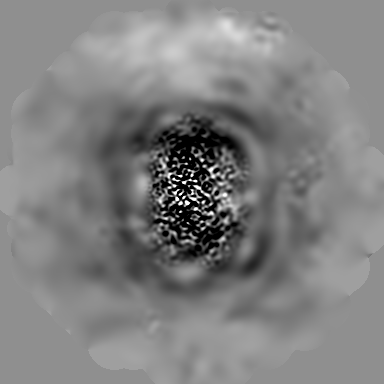
| Projections & Slices |
| ||||||||||||
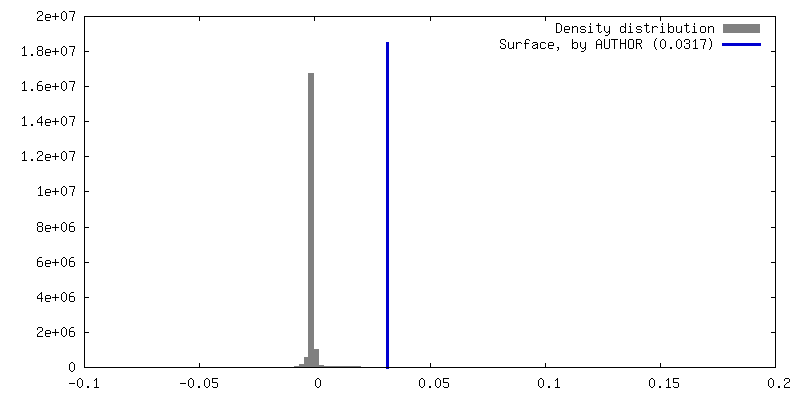
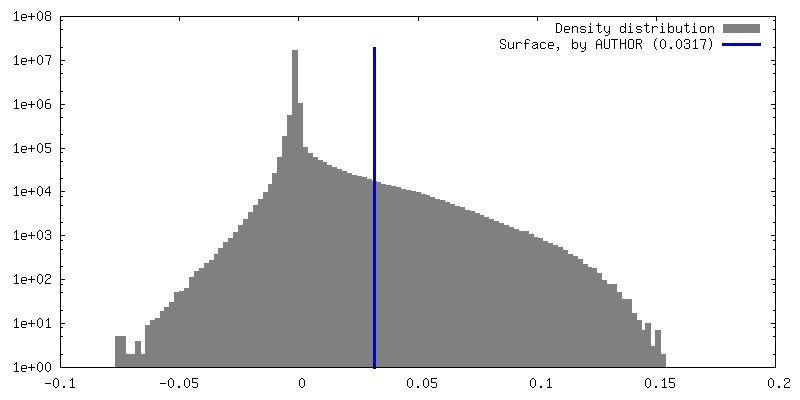
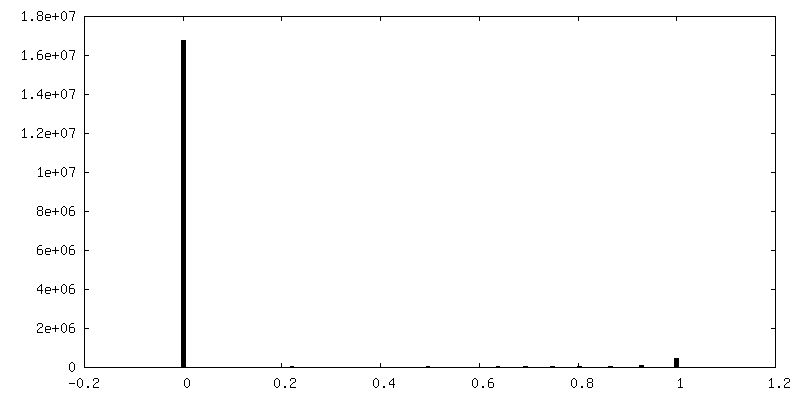
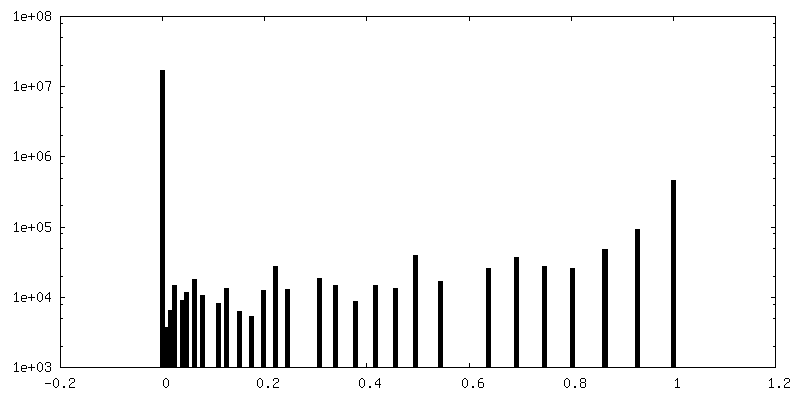
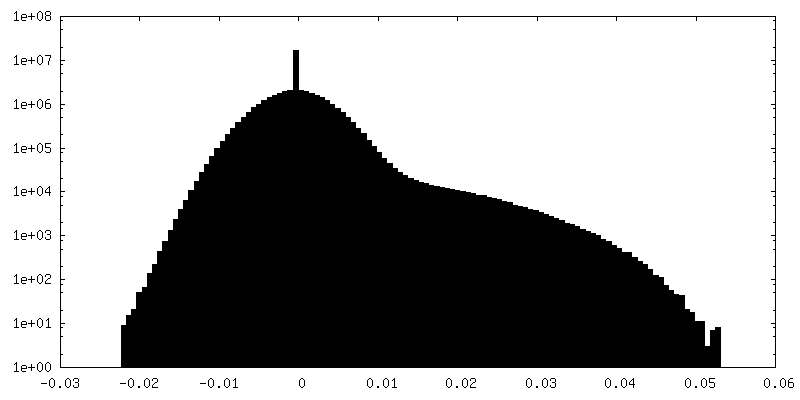
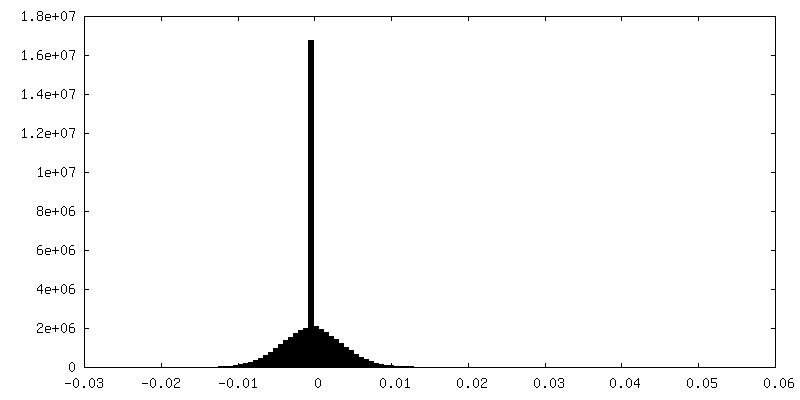
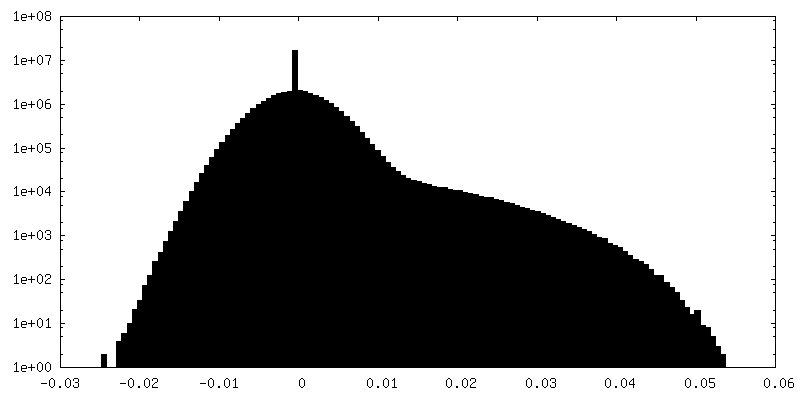
| Density Histograms |






-Half map: CryoEM structure of GroEL bound to non-native Rubisco.
| File | emd_15939_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM structure of GroEL bound to non-native Rubisco. | ||||||||||||
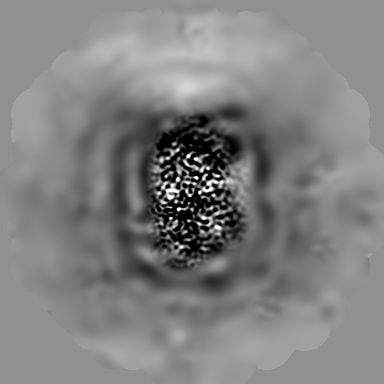
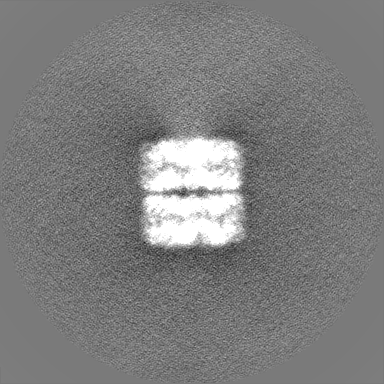
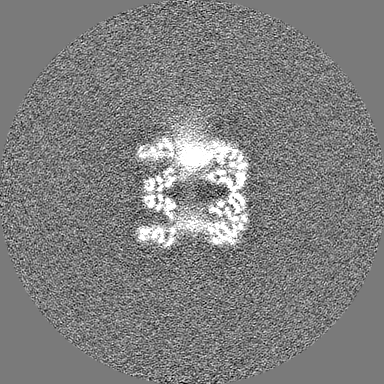
| Projections & Slices |
| ||||||||||||
| Density Histograms |



-Half map: CryoEM structure of GroEL bound to non-native Rubisco.
| File | emd_15939_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM structure of GroEL bound to non-native Rubisco. | ||||||||||||
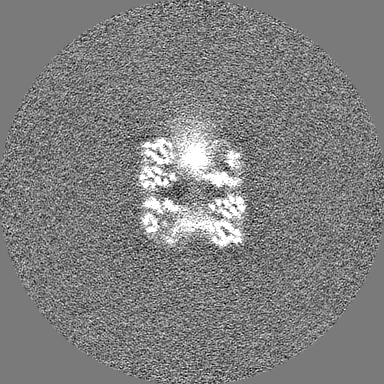
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : GroEL
| Entire | Name: GroEL |
|---|---|
| Components |
|
-Supramolecule #1: GroEL
| Supramolecule | Name: GroEL / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) / Strain: K12 |
| Molecular weight | Theoretical: 802 KDa |
-Macromolecule #1: Chaperonin GroEL
| Macromolecule | Name: Chaperonin GroEL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO / EC number: chaperonin ATPase |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) / Strain: K12 |
| Molecular weight | Theoretical: 57.260504 KDa |
| Recombinant expression | Organism: Escherichia coli BL21(DE3) (bacteria) |
| Sequence | String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV ...String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV GKEGVITVED GTGLQDELDV VEGMQFDRGY LSPYFINKPE TGAVELESPF ILLADKKISN IREMLPVLEA VA KAGKPLL IIAEDVEGEA LATLVVNTMR GIVKVAAVKA PGFGDRRKAM LQDIATLTGG TVISEEIGME LEKATLEDLG QAK RVVINK DTTTIIDGVG EEAAIQGRVA QIRQQIEEAT SDYDREKLQE RVAKLAGGVA VIKVGAATEV EMKEKKARVE DALH ATRAA VEEGVVAGGG VALIRVASKL ADLRGQNEDQ NVGIKVALRA MEAPLRQIVL NCGEEPSVVA NTVKGGDGNY GYNAA TEEY GNMIDMGILD PTKVTRSALQ YAASVAGLMI TTECMVTDLP KNDAADLGAA GGMGGMGGMG GMM UniProtKB: Chaperonin GroEL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2.7 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 80 % / Chamber temperature: 293 K / Instrument: SPOTITON Details: The grid was prepared using a chameleon (SPT Labtech).. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.4000000000000001 µm |
| Image recording | Film or detector model: GATAN K2 BASE (4k x 4k) / Number grids imaged: 2 / Average electron dose: 40.2 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: PDB ENTRY PDB model - PDB ID: |
|---|---|
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final 3D classification | Number classes: 8 / Avg.num./class: 25000 / Software - Name: RELION (ver. 3.1) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final reconstruction | Number classes used: 2 / Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 4.4 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) / Number images used: 65453 |
| FSC plot (resolution estimation) | 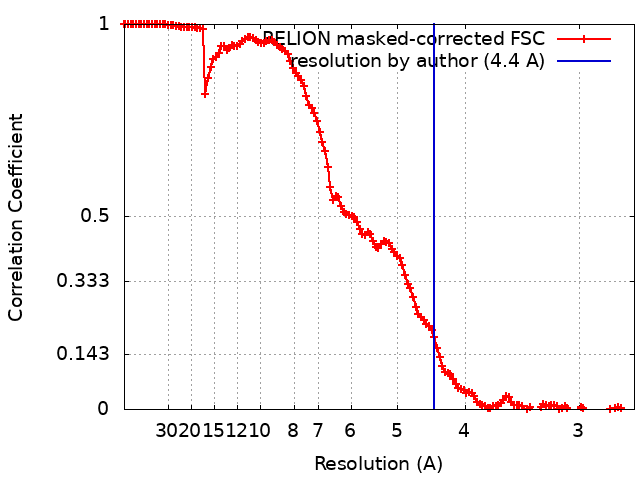 |

