 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
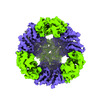
Yorodumi- PDB-8umr: T33-ml35 Assembly Intermediate - Designed Tetrahedral Protein Cag... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8umr | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | T33-ml35 Assembly Intermediate - Designed Tetrahedral Protein Cage Using Machine Learning Algorithms | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords |  DE NOVO PROTEIN / Nanohedra / protein cage / tetrahedral / de novo protein interface / machine learning / two components / ProteinMPNN / nanoparticle DE NOVO PROTEIN / Nanohedra / protein cage / tetrahedral / de novo protein interface / machine learning / two components / ProteinMPNN / nanoparticle | |||||||||
| Biological species | synthetic construct (others) | |||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 4.42 Å | |||||||||
 Authors Authors | Castells-Graells, R. / Meador, K. / Sawaya, M.R. / Yeates, T.O. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2024 Title: A suite of designed protein cages using machine learning and protein fragment-based protocols. Authors: Kyle Meador / Roger Castells-Graells / Roman Aguirre / Michael R Sawaya / Mark A Arbing / Trent Sherman / Chethaka Senarathne / Todd O Yeates / Abstract: Designed protein cages and related materials provide unique opportunities for applications in biotechnology and medicine, but their creation remains challenging. Here, we apply computational ...Designed protein cages and related materials provide unique opportunities for applications in biotechnology and medicine, but their creation remains challenging. Here, we apply computational approaches to design a suite of tetrahedrally symmetric, self-assembling protein cages. For the generation of docked conformations, we emphasize a protein fragment-based approach, while for sequence design of the de novo interface, a comparison of knowledge-based and machine learning protocols highlights the power and increased experimental success achieved using ProteinMPNN. An analysis of design outcomes provides insights for improving interface design protocols, including prioritizing fragment-based motifs, balancing interface hydrophobicity and polarity, and identifying preferred polar contact patterns. In all, we report five structures for seven protein cages, along with two structures of intermediate assemblies, with the highest resolution reaching 2.0 Å using cryo-EM. This set of designed cages adds substantially to the body of available protein nanoparticles, and to methodologies for their creation. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule:  MolmilJmol/JSmol MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8umr.cif.gz | 399.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8umr.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 8umr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/um/8umrftp://data.pdbj.org/pub/pdb/validation_reports/um/8umr | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 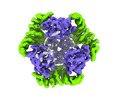 42382MC  8uf0C  8ui2C  8ujaC  8ukmC  8umpC  8un1C M: map data used to model this data C: citing same article ( |
|---|

-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 13557.165 Da / Num. of mol.: 9 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Plasmid: pSAM / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): LOBSTR Escherichia coli BL21(DE3) (bacteria) / Strain (production host): LOBSTR#2: Protein | Mass: 14324.456 Da / Num. of mol.: 12 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Plasmid: pSAM / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): LOBSTR |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: T33-ml35 Assembly Intermediate - Designed Tetrahedral Protein Cage Using Machine Learning Type: COMPLEX / Entity ID: all / Source: RECOMBINANT |
|---|---|
| Molecular weight | Value: 0.4 MDa / Experimental value: YES |
| Source (natural) | Organism: synthetic construct (others) |
| Source (recombinant) | Organism: Escherichia coli BL21(DE3) (bacteria) / Strain: LOBSTR |
| Buffer solution | pH: 7.5 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Grid material: COPPER / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil R2/1 |
| Vitrification | Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 291 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source: FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 2500 nm / Nominal defocus min: 500 nm / Cs: 2.7 mm |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Electron dose: 47.5 e/Å2 / Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 4.42 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 66561 / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT | ||||||||||||||||||||||||
| Refine LS restraints |
|
