 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7oz3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | S. agalactiae BusR in complex with its busA-promotor DNA | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords |  DNA BINDING PROTEIN / Repressor / complex / GntR / Transcription DNA BINDING PROTEIN / Repressor / complex / GntR / Transcription | |||||||||
| Function / homology |  Function and homology information Function and homology informationmonoatomic cation transmembrane transporter activity / potassium ion transport / DNA-binding transcription factor activity Similarity search - Function | |||||||||
| Biological species |  Streptococcus agalactiae (bacteria) Streptococcus agalactiae (bacteria) | |||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 4.46 Å | |||||||||
 Authors Authors | Bandera, A.M. / Witte, G. | |||||||||
| Funding support |  Germany, 2items Germany, 2items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2021 Title: BusR senses bipartite DNA binding motifs by a unique molecular ruler architecture. Authors: Adrian M Bandera / Joseph Bartho / Katja Lammens / David Jan Drexler / Jasmin Kleinschwärzer / Karl-Peter Hopfner / Gregor Witte / Abstract: The cyclic dinucleotide second messenger c-di-AMP is a major player in regulation of potassium homeostasis and osmolyte transport in a variety of bacteria. Along with various direct interactions with ...The cyclic dinucleotide second messenger c-di-AMP is a major player in regulation of potassium homeostasis and osmolyte transport in a variety of bacteria. Along with various direct interactions with proteins such as potassium channels, the second messenger also specifically binds to transcription factors, thereby altering the processes in the cell on the transcriptional level. We here describe the structural and biochemical characterization of BusR from the human pathogen Streptococcus agalactiae. BusR is a member of a yet structurally uncharacterized subfamily of the GntR family of transcription factors that downregulates transcription of the genes for the BusA (OpuA) glycine-betaine transporter upon c-di-AMP binding. We report crystal structures of full-length BusR, its apo and c-di-AMP bound effector domain, as well as cryo-EM structures of BusR bound to its operator DNA. Our structural data, supported by biochemical and biophysical data, reveal that BusR utilizes a unique domain assembly with a tetrameric coiled-coil in between the binding platforms, serving as a molecular ruler to specifically recognize a 22 bp separated bipartite binding motif. Binding of c-di-AMP to BusR induces a shift in equilibrium from an inactivated towards an activated state that allows BusR to bind the target DNA, leading to transcriptional repression. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7oz3.cif.gz | 196.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7oz3.ent.gz | 152.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7oz3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oz/7oz3ftp://data.pdbj.org/pub/pdb/validation_reports/oz/7oz3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  13119MC  7b5tC  7b5uC  7b5wC  7b5yC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 23880.160 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus agalactiae (bacteria)Gene: BM110_ORF1201, AX245_01365, C6N10_09995, F5043_05515, GD434_05225, RDF_1124 Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: K0JNC6#2: DNA chain | | Mass: 46903.086 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Streptococcus agalactiae (bacteria)#3: DNA chain | | Mass: 46903.062 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Streptococcus agalactiae (bacteria)#4: Chemical | 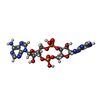  Mass: 658.412 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24N10O12P2 / Feature type: SUBJECT OF INVESTIGATION Mass: 658.412 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H24N10O12P2 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: transcriptional repressor BusR bound to target DNA / Type: COMPLEX / Entity ID: #1-#3 / Source: MULTIPLE SOURCES | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.122 MDa | |||||||||||||||
| Source (natural) | Organism: Streptococcus agalactiae (bacteria) | |||||||||||||||
| Buffer solution | pH: 6.5 / Details: degassed, filtered | |||||||||||||||
| Buffer component |
| |||||||||||||||
| Specimen | Conc.: 0.6 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | |||||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 200 divisions/in. / Grid type: Quantifoil R2/1 | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Humidity: 95 % / Chamber temperature: 283 K Details: 0.05% beta-octyl glycoside added prior to plunge freezing |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source: FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELDBright-field microscopy |
| Image recording | Electron dose: 45 e/Å2 / Detector mode: COUNTING / Film or detector model: GATAN K2 SUMMIT (4k x 4k) |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| ||||||||||||||||||||||||||||||||||||
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 4.46 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 112451 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||
| Atomic model building | B value: 177 / Protocol: FLEXIBLE FIT / Space: REAL Details: RCK_C domains were build by rigid body fit of the model 7B5U. The flexible linker between RCK_C domain and coiled-coil domain were build de novo. | ||||||||||||||||||||||||||||||||||||
| Atomic model building |
| ||||||||||||||||||||||||||||||||||||
| Refinement | Cross valid method: NONE Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 134.6 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
