 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lcu: Polylysine Induces an Antiparallel Actin Dimer that Nucleates Fil... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lcu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Polylysine Induces an Antiparallel Actin Dimer that Nucleates Filament Assembly: Crystal Structure at 3.5 A Resolution | ||||||
 Components Components | Actin, alpha skeletal muscle | ||||||
 Keywords Keywords | CONTRACTILE PROTEIN / Structural protein / Muscle protein | ||||||
| Function / homology |  Function and homology information Function and homology informationcytoskeletal motor activator activity / tropomyosin binding / mesenchyme migration / troponin I binding / myosin heavy chain binding / filamentous actin / actin filament bundle / skeletal muscle thin filament assembly / striated muscle thin filament / actin filament bundle assembly ...cytoskeletal motor activator activity / tropomyosin binding / mesenchyme migration / troponin I binding / myosin heavy chain binding / filamentous actin / actin filament bundle / skeletal muscle thin filament assembly / striated muscle thin filament / actin filament bundle assembly / skeletal muscle myofibril / actin monomer binding / skeletal muscle fiber development / stress fiber / titin binding / actin filament polymerization / filopodium / actin filament / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement / calcium-dependent protein binding / lamellipodium / cell body / hydrolase activity / protein domain specific binding / calcium ion binding / positive regulation of gene expression / magnesium ion binding / ATP binding / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.5 Å | ||||||
 Authors Authors | Bubb, M.R. / Govindasamy, L. / Yarmola, E.G. / Vorobiev, S.M. / Almo, S.C. / Somasundaram, T. / Chapman, M.S. / Agbandje-Mckenna, M. / Mckenna, R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Polylysine induces an antiparallel actin dimer that nucleates filament assembly: crystal structure at 3.5-A resolution. Authors: Bubb, M.R. / Govindasamy, L. / Yarmola, E.G. / Vorobiev, S.M. / Almo, S.C. / Somasundaram, T. / Chapman, M.S. / Agbandje-Mckenna, M. / Mckenna, R. #1: Journal: J.Biol.Chem. / Year: 2000Title: Effects of Jasplakinolide on the Kinetics of Actin Polymerization. AN EXPLANATION FOR CERTAIN IN VIVO OBSERVATIONS Authors: Bubb, M.R. / Spector, I. / Beyer, B.B. / Fosen, K.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lcu.cif.gz | 164.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lcu.ent.gz | 126.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1lcu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1lcu_validation.pdf.gz | 665.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1lcu_full_validation.pdf.gz | 712.9 KB | Display | |
| Data in XML | 1lcu_validation.xml.gz | 22.9 KB | Display | |
| Data in CIF | 1lcu_validation.cif.gz | 32.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lc/1lcuftp://data.pdbj.org/pub/pdb/validation_reports/lc/1lcu | HTTPS FTP |
-Related structure data
| Related structure data |  1ijjC  1esvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 41344.160 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: Muscle / Source: (natural) |
|---|
-Non-polymers , 5 types, 95 molecules 


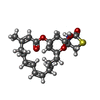

| #2: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical |  Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#4: Chemical |  Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#5: Chemical |  Mass: 421.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H31NO5S / Comment: toxin*YM Mass: 421.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H31NO5S / Comment: toxin*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 8 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.01 Å3/Da / Density % sol: 69.33 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: Ammonium sulfate, MgCl2, Imidazole, ATP, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion / pH: 6.7 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Apr 4, 2000 / Details: OSMIC MIRROR |
| Radiation | Monochromator: Cu / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→30 Å / Num. obs: 15407 / % possible obs: 88.8 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Rmerge(I) obs: 0.115 |
| Reflection shell | Highest resolution: 3.5 Å / Rmerge(I) obs: 0.115 / Num. unique all: 15407 / % possible all: 88.8 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. measured all: 101490 |
| Reflection shell | *PLUS Rmerge(I) obs: 0.249 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ESV Resolution: 3.5→30 Å / σ(F): 1 / σ(I): 1 / Stereochemistry target values: Brunger & Adams
| ||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Highest resolution: 3.5 Å
| ||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / % reflection Rfree: 5 % / Rfactor all: 0.196 / Rfactor obs: 0.193 / Rfactor Rfree: 0.266 / Rfactor Rwork: 0.193 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
