 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8267 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
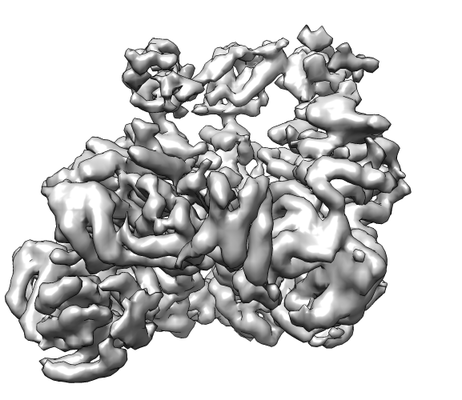
| Title | CryoEM Reconstruction of Hsp104 Hexamer | |||||||||||||||
 Map data Map data | Hexamer Reconstruction | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | Hsp104 / AAA+ protein / CHAPERONE | |||||||||||||||
| Function / homology |  Function and homology information Function and homology information: / TRC complex / protein folding in endoplasmic reticulum / cellular heat acclimation / post-translational protein targeting to endoplasmic reticulum membrane / stress granule disassembly / : / protein unfolding / nuclear periphery / ADP binding ...: / TRC complex / protein folding in endoplasmic reticulum / cellular heat acclimation / post-translational protein targeting to endoplasmic reticulum membrane / stress granule disassembly / : / protein unfolding / nuclear periphery / ADP binding / unfolded protein binding / protein-folding chaperone binding / cellular response to heat / protein refolding / ATP hydrolysis activity / ATP binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||||||||
| Biological species |  | |||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 5.64 Å | |||||||||||||||
 Authors Authors | Yokom AL / Gates SN | |||||||||||||||
| Funding support |  United States, 4 items United States, 4 items
| |||||||||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2016 Title: Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Authors: Adam L Yokom / Stephanie N Gates / Meredith E Jackrel / Korrie L Mack / Min Su / James Shorter / Daniel R Southworth / Abstract: Hsp104, a conserved AAA+ protein disaggregase, promotes survival during cellular stress. Hsp104 remodels amyloids, thereby supporting prion propagation, and disassembles toxic oligomers associated ...Hsp104, a conserved AAA+ protein disaggregase, promotes survival during cellular stress. Hsp104 remodels amyloids, thereby supporting prion propagation, and disassembles toxic oligomers associated with neurodegenerative diseases. However, a definitive structural mechanism for its disaggregase activity has remained elusive. We determined the cryo-EM structure of wild-type Saccharomyces cerevisiae Hsp104 in the ATP state, revealing a near-helical hexamer architecture that coordinates the mechanical power of the 12 AAA+ domains for disaggregation. An unprecedented heteromeric AAA+ interaction defines an asymmetric seam in an apparent catalytic arrangement that aligns the domains in a two-turn spiral. N-terminal domains form a broad channel entrance for substrate engagement and Hsp70 interaction. Middle-domain helices bridge adjacent protomers across the nucleotide pocket, thus explaining roles in ATP hydrolysis and protein disaggregation. Remarkably, substrate-binding pore loops line the channel in a spiral arrangement optimized for substrate transfer across the AAA+ domains, thereby establishing a continuous path for polypeptide translocation. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8267.map.gz | 10.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8267-v30.xmlemd-8267.xml | 13 KB 13 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8267_fsc.xml | 5.2 KB | Display | FSC data file |
| Images |  emd_8267.png emd_8267.png | 89.2 KB | ||
| Filedesc metadata | emd-8267.cif.gz | 6.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8267ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8267 http://ftp.pdbj.org/pub/emdb/structures/EMD-8267ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8267 | HTTPS FTP |
-Validation report
| Summary document | emd_8267_validation.pdf.gz | 546.7 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_8267_full_validation.pdf.gz | 546.3 KB | Display | |
| Data in XML | emd_8267_validation.xml.gz | 7.6 KB | Display | |
| Data in CIF | emd_8267_validation.cif.gz | 9.8 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-8267ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-8267 | HTTPS FTP |
-Related structure data
| Related structure data |  5kneMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_8267.map.gz / Format: CCP4 / Size: 11.4 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Hexamer Reconstruction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Hexamer Complex of Hsp104
| Entire | Name: Hexamer Complex of Hsp104 |
|---|---|
| Components |
|
-Supramolecule #1: Hexamer Complex of Hsp104
| Supramolecule | Name: Hexamer Complex of Hsp104 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Heat shock protein 104
| Macromolecule | Name: Heat shock protein 104 / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 95.967398 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: QFTERALTIL TLAQKLASDH QHPQLQPIHI LAAFIETPED GSVPYLQNLI EKGRYDYDLF KKVVNRNLVR IPQQQPAPAE ITPSYALGK VLQDAAKIQK QQKDSFIAQD HILFALFNDS SIQQIFKEAQ VDIEAIKQQA LELRGNTRID SRGADTNTPL E YLSKYAID ...String: QFTERALTIL TLAQKLASDH QHPQLQPIHI LAAFIETPED GSVPYLQNLI EKGRYDYDLF KKVVNRNLVR IPQQQPAPAE ITPSYALGK VLQDAAKIQK QQKDSFIAQD HILFALFNDS SIQQIFKEAQ VDIEAIKQQA LELRGNTRID SRGADTNTPL E YLSKYAID MTEQARQGKL DPVIGREEEI RSTIRVLARR IKSNPCLIGE PGIGKTAIIE GVAQRIIDDD VPTILQGAKL FS LDLAALT AGAKYKGDFE ERFKGVLKEI EESKTLIVLF IDEIHMLMGN GKDDAANILK PALSRGQLKV IGATTNNEYR SIV EKDGAF ERRFQKIEVA EPSVRQTVAI LRGLQPKYEI HHGVRILDSA LVTAAQLAKR YLPYRRLPDS ALDLVDISCA GVAV ARDSK PEELDSKERQ LQLIQVEIKA LERDEDADST TKDRLKLARQ KEASLQEELE PLRQRYNEEK HGHEELTQAK KKLDE LENK ALDAERRYDT ATAADLRYFA IPDIKKQIEK LEDQVAEEER RAGANSMIQN VVDSDTISET AARLTGIPVK KLSESE NEK LIHMERDLSS EVVGQMDAIK AVSNAVRLSR SGLANPRQPA SFLFLGLSGS GKTELAKKVA GFLFNDEDMM IRVDCSE LS EKYAVSKLLG TTAGYVGYDE GGFLTNQLQY KPYSVLLFDE VEKAHPDVLT VMLQMLDDGR ITSGQGKTID CSNCIVIM T SNLGAEFINS QQGSKIQEST KNLVMGAVRQ HFRPEFLNRI SSIVIFNKLS RKAIHKIVDI RLKEIEERFE QNDKHYKLN LTQEAKDFLA KYGYSDDMGA RPLNRLIQNE ILNKLALRIL KNEIKDKETV NV UniProtKB: Heat shock protein 104 |
-Macromolecule #2: PHOSPHOAMINOPHOSPHONIC ACID-ADENYLATE ESTER
| Macromolecule | Name: PHOSPHOAMINOPHOSPHONIC ACID-ADENYLATE ESTER / type: ligand / ID: 2 / Number of copies: 11 / Formula: ANP |
|---|---|
| Molecular weight | Theoretical: 506.196 Da |
| Chemical component information |  ChemComp-ANP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.7 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: C-flat / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Support film - Film thickness: 2 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV Details: Plunged into liquid ethane (FEI VITROBOT MARK IV). |
| Details | Clean protein sample incubated with AMP-PNP |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 2-40 / Number grids imaged: 2 / Number real images: 3661 / Average exposure time: 0.2 sec. / Average electron dose: 1.6 e/Å2 Details: Movies contained 38 of 40 frames from an 8 sec exposure (200 milliseconds per frame). |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Details | Backbone atoms fit into reconstruction density. |
|---|---|
| Output model | PDB-5kne: |
