The program gmconvert converts both a 3D density map and an atomi model into GMM (gaussian mixture model). EM (expectation maximization) algorithm is employed for covertion into GMM. The program gmconvert also has many other useful functions to handle GMM.
The source code of gmconvert is written in C assuming the compiler "gcc" in Linux environment.
After you download the file gmconvert-src-[date].tar.gz, just type following commands:
tar zxvf gmconvert-src-[date].tar.gz cd src makeThen you will find the execute file
gmconvert in the upper directory (../src).
- 'A2G' (atom-to-gauss): [PDB atom] --> [GMM]
gmconvert A2G -ipdb [PDBfile] -ogmm [GMMfile] -ng [Number of Gaussian functions]
- 'V2G' (voxel-to-gauss): [3D density map] --> [GMM]
gmconvert V2G -imap [CCP4/MRC map file] -ogmm [GMMfile] -ng [Number of Gaussian functions]
- 'A2V' (atom-to-voxel): [PDB atom] --> [3D density map]
gmconvert A2V -ipdb [PDBfile] -omap [CCP4/MRC map file] -reso [resolution]
- 'G2S' (gauss-to-surface):[GMM] --> [Wireframe or surface in PDB]
gmconvert G2S -igmm [GMM] -gw [grid_width] -opdb [PDB]
- 'G2V' (gauss-to-voxel) :[GMM] --> [3D density map]
gmconvert G2V -igmm [GMM] -omap [CCP4/MRC map file] -gw [grid_width]
- 'G2E' (gauss-to-ellipsoid):[GMM] --> [VRML file for ellipsoids]
gmconvert -igmm [GMM] -oewrl [VRML file]
- 'V2S' (voxel-to-surface): [3D density map] --> [Wireframe surface in PDB]
gmconvert V2S -imap [CCP4/MRC mapfile] -opdb [PDB] -gw [grid_width]
- 'VcmpG' (voxel-compare with-gauss): Comparison between [GMM] and [3D density map]
gmconvert VcmpG -igmm [GMMfile] -imap [CCP4/MRC map file]
- 'GcmpG' (gauss-compare_with-gauss):Comparison between [GMM] and another [GMM]
gmconvert GcmpG -igmm [GMMfile] -igmm2 [GMMfile2]
- 'A2I' (atom-to-2Dimage): Make a projection image from atomic model
gmconvert A2I -ipdb [PDBfile] -o2Dpgm [projection 2D image (*.pgm)]
- 'G2I' (atom-to-2Dimage): Make a projection image from GMM.
gmconvert G2I -igmm [PDBfile] -o2Dpgm [projection 2D image (*.pgm)]
- 'GbyA (gauss-by-atom): Transform [GMM] by comparison bwn [PDB] and target [PDB]. For test for symmetric complex.
gmconvert GbyA -ipdb [PDB] -itpdb [targetPDB] -igmm [GMM] -ogmm [target GMM]
- Show the options for the [MODE]
gmconvert [MODE] -h
- Show all the options
gmconvert -h
- Basic Usage
To convert a 3D density map into a GMM, a basic command is as follows:
gmconvert V2G -imap [CCP4/MRC map file] -ogmm [GMMfile] -cutoff [threshold for density map] -ng [Number of Gaussian functions]
The threshold density is imporant to denoise the density map. It can be assigned by the options-cutoffor-cutsd.-cutoff: if density < [-cutoff], it is regarded as zero density. [-1.000000]
If a density of a voxel is less than the-cutoffvalue, its density is assigned as zero. After that, the voxel is regarded as the place where no voxel exists. Only positive-cutoffvalue is meaningful. The negative-cutoffvalue will be ignored. If you use the density map from EMDB, we recommend to use the author-recommended contour level.-cutsd: if density < MEAN + [-cutsd]*SD, it is regarded as zero density. [3.000000]
If you do not know the proper threshold value, the statistics of the density map will help you. If the option-cutsdis assigned, the threshold value is [MEAN of density] +[-cutoff]* [SD of density]. Only positive-cutsdvalue is meaningful. The negative-cutsdvalue will be ignored. If both-cutoffand-cutsdare positive, the option-cutoffhas a priority.
A following command is for converting the density map EMDB-2190 into a GMM with Ngauss = 10, using the threshold value 0.066, which is taken from the author-recommend contour level:
gmconvert V2G -imap emd_2190.map -cutoff 0.0666 -ng 10 -ogmm emd_2190_ng10.gmm
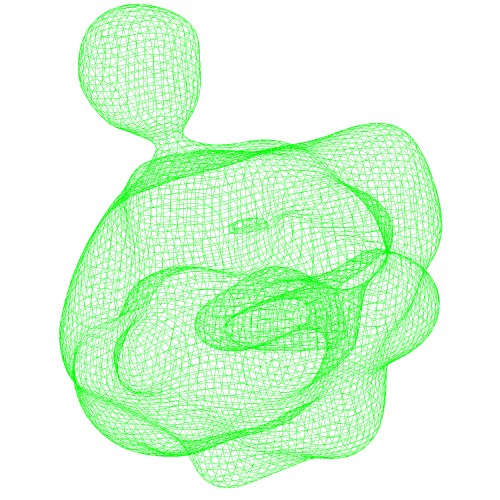
The generated GMM can be visually checked by various formats. The simplest way is just open the generated "*.gmm" file as the PDB file by molecular graphics program. Then, the centers of Gaussian functions are shown.The isocontour surface model in VRML format (
*.wrl) can be generated by thegmconvertby a following command:gmconvert G2S -igmm emd_2190_ng10.gmm -owrl emd_2190_ng10_surface.wrl
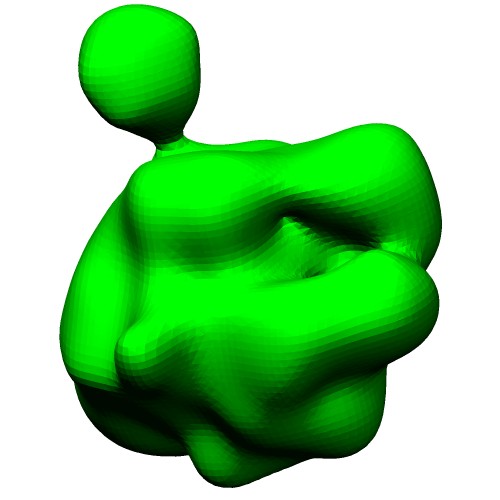
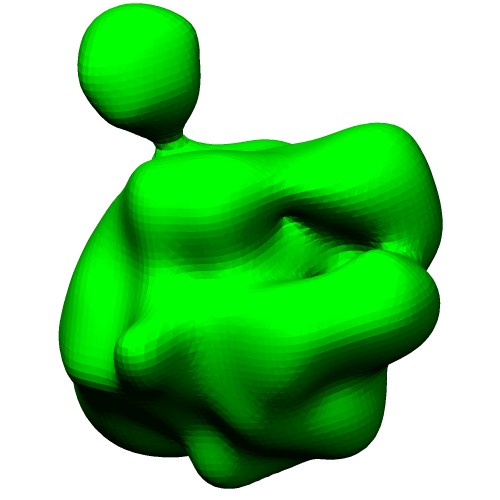
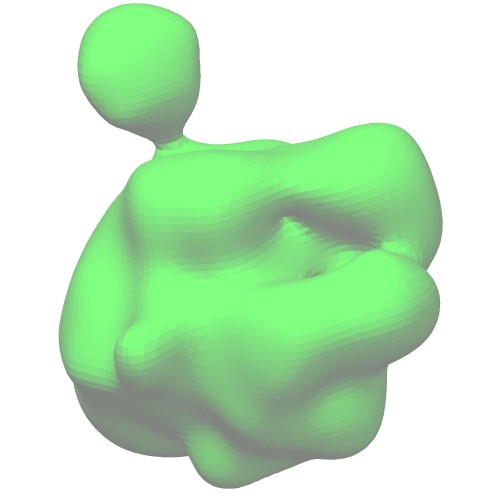
The ellipsoid model in VRML format (*.wrl) can be generated by thegmconvertby a following command:gmconvert G2E -igmm emd_2190_ng10.gmm -oewrl emd_2190_ng10_ellipsoid.wrl
These VRML files (*.wrl) can visualised by the program UCSF Chimera.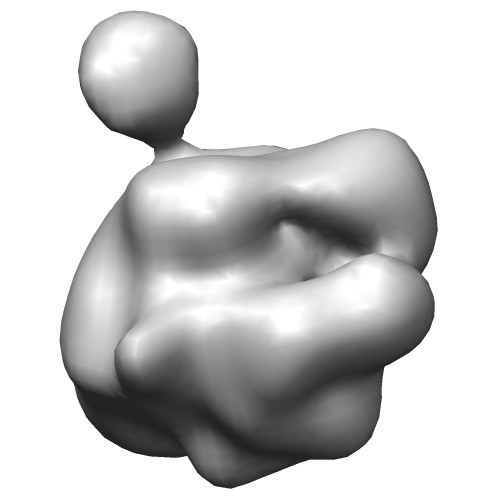

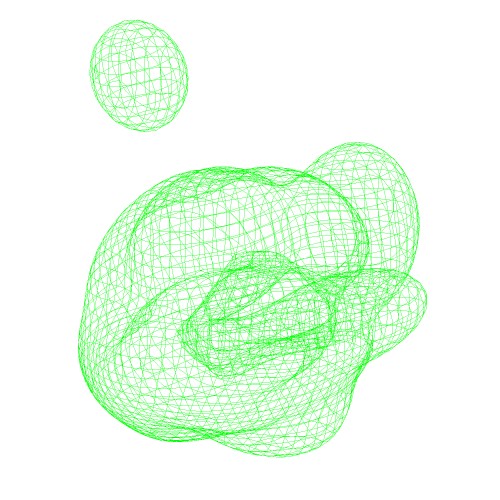

original map (EMD-2190)
with threshold 0.0666Centers of GMM
(emd_2190_ng10.gmm)Wireframe of GMM isocontour surface
(emd_2190_ng10_surface.wrl)Ellipsoids of GMM
(emd_2190_ng10_ellipsoid.wrl) - Algorithms of fitting GMM with the density map
The program
gmconvertemploys the EM (expectarion-maximization) algorithm and down-sampling algorithm for fittting GMM with the given density map.
NOTE: The optionoption -emalgdefault or not name explanation -emalg WPWeighted point-input GMM (WP-input GMM) Each voxel is represented by the center point with weights of density -emalg Gdefault Gaussian-input GMM (G-input GMM) Each voxel is represented by the isotropic Gaussian function with variance = [grid_width]/12. -emalg DDown-sampled Gaussian function (DSG) Neighboring voxles are merged into one Gaussian functions. The option -fdsmapis requried.-emalg DGDown-sampled Gaussian-input GMM (DSG-input GMM) Down-sampled Gaussian functions are used for the input of G-input GMM. The option -fdsmapis requried.-emalg Pdoes not work for the density map.The convergence of the EM algorithm will be determined mainly by following two parameters.
-nr: Number of repeat for EM [10000]-cv: Convergence threshold for dParam for the EM algorithm [0.001000]
Especially, the option
-nrstrongly affects the computation time. The default value is-nr 10000. If less number is assigned, for example,-nr 1000, the program may finish before 1/10 computation time, although the final likelihood value may not be fully converged. - For the map with large number of voxels
It often requires large amount of time (more than hours) to convert the map with large number of voxels (such as 5123 voxles) into GMM. We recommend following two ways to the fast conversion of these large map.
- Down-sampled Gaussian functions (DSG) :
-emalg DThe first way is employing the down-sampled Gaussian functions (DSG). This is merging neighboring voxels into one anisotropic Gaussian functions to generate GMM. An command example is as follows:
gmconvert V2G -imap [map file] -ogmm [GMMfile] -cutoff [threshold] -emalg D -fdsmap [down-sampling factor]
Number of neighboring voxels can be controlled by the option
-fdsmap.[fdsmap]3 voxels are merged into one Gaussian function. For example, if the option-fdsmap 2is assigned, 2 x 2 x 2 = 8 voxels are merged into one Gaussian function. In the case of-emalg D, the number of Gaussian functions of the GMM cannot be assigened by the option-ng, it depends on number of foreground voxels in the original map and the down-sampling factor-fdsmap.Instead of the option
-fdsmap, the width of downsampled voxel can be assigned using the option-wdsmap.gmconvert V2G -imap [map file] -ogmm [GMMfile] -cutoff [threshold] -emalg D -wdsmap [width of down-sampled voxel]
The width of downsampled voxel-wdsmapis used to decide the value of-fdsmap;[fdsmap] = floor([wdsmap]/[grid_width]). For example, if the grid width of the given map is 0.82 angstrom, and the width-wdsmap 2.0, then the factor-fdsmapis 2.Command examples using emd_6643 (5123 voxels) are shown as follows:
gmconvert V2G -imap emd_6643.map -cutoff 0.018 -emalg D -fdsmap 16 -ogmm emd_6643_D_df16.gmm
gmconvert G2E -igmm emd_6643_D_df16.gmm -cutoff 0.018 -oewrl emd_6643_D_df16_ellipsoid.wrl -elcol W -elRGBT 0:0:0:0
The genration of DSG is quite fast, because it does not require any iterative calulation. The DSG is suitable for generating a GMM with a large number of Gaussian functoins (such as > 1000) for a high-resolution density map. - Down-sampled Gaussian-input GMM (DSG-input GMM) :
-emalg DGThe second way is employing the DSG-input GMM. First, a down-sampled Gaussian functoins (DSG) is generated, then the Gaussian-input GMM alrogithm is applied to the genrated DSG. The DSG-input GMM is suitable for generating a GMM with a small number of Gaussian functoins (such as < 100). An command example is as follows:
gmconvert V2G -imap [map file] -ogmm [GMMfile] -cutoff [threshold] -emalg DG -fdsmap [down-sampling factor] -ng [Ngauss]
Note that the option-ngis required for the case of the optionDG. Command examples using emd_6643 (5123 voxels) are shown as follows:gmconvert V2G -imap emd_6643.map -cutoff 0.018 -emalg DG -fdsmap 16 -ng 10 -ogmm emd_6643_DG_df16_ng10.gmm
gmconvert G2E -igmm emd_6643_DG_df16_ng10.gmm -oewrl emd_6643_DG_df16_ng10_ellipsoid.wrl -elcol W -elRGBT 0:0:0:0



Original map (EMD-6643)
threshold density:0.018
Number of voxels 5123
Number of foreground voxels:2057291Down-sampled Gaussian functions (DSG)
by ellipsoidal representation.
(emd_6643_D_df16_ellipsoid.wrl)
Number of Gaussian functions: 2995DSG-input GMM
by ellipsoidal representation.
(emd_6643_DG_df16_ng10_ellipsoid.wrl)
Number of Gaussian functions: 10 - Gaussian-input with down sampling (G) :
-emalg Gwith-fdsmapThis is not our recommended way. The Gaussian-input GMM algorithm can also use the down-sampling using
-fdsmap. Different from the DG-input GMM (-emalg DG), the G-input GMM first converts the original map into a down sampled map with-fdsmap, and regards the map as a set of "isotropic" Gaussian functions, not anistropic ones. An example is as follows:gmconvert V2G -imap emd_6643.map -cutoff 0.018 -emalg G -fdsmap 16 -ng 10 -ogmm emd_6643_G_df16_ng10.gmm -omapds emd_6643_df16.map
The input isotropic Gaussian functions can be seen as follows:gmconvert V2G -imap emd_6643_df16.map -cutoff 0.018 -emalg O -ogmm emd_6643_df16.gmm gmconvert G2E -igmm emd_6643_df16.gmm -oewrl emd_6643_df16_ellipsoid.wrl -elcol W -elRGBT 0:0:0:0
As you see, the istropic Gaussian functions from the downsampled map less conserve the features of the original map than the anisotroic GMM. It does not conserve the covariance matrix of the original map.

Isotropic Gaussian functions
from down-sampled map(DSG)
by ellipsoidal representation.
(emd_6643_df16_ellipsoid.wrl)
Number of Gaussian functions: 2995
- Down-sampled Gaussian functions (DSG) :
- Other Options
-maxsize: allowed max voxel size of each axis. If over, downsample to isotropic gauss (-emalg G/WP) or anisotropic gauss (-emalg D). [-1]-fdsmap: downsampling factor (2,3,4,...) for -emalg G/W or -emalg D or -emalg DG. [1]-vargrd: Variance type for grid points (for -emalg G or T). 'G':var = ww2var * grid_width * grid_width, 'R': var = (resolution/2.0)^2.[G]-ww2var: Constant for variance = Const * grid_width*grid_width for emalg G. Default is 1/12 [0.083333].-resogrd: Resolution for grid point (for -emalg G -vargrd R) [0.000000].-resoblurgrd: Resolution of grid point for blurring the map. [0.000000].-ogmm: Output Gaussian File (*.gmm) []-ng: Number of Gaussian functions for GMM [1]-emalg: type for EM algorithm. 'P'oint-input,'WP'eighted_point-input 'G'aussian-input (isotropic) 'O':1-to-1 atom/grid pnts : 'D':Down-sampled gaussians 'DG':Down-sampled gaussian-input GMM [G] : note: 'D' and 'DG' is only for map (V2G)-I: Initialization of GMM. 'K'-means, 'R'andom 'O':one-to-one_atom/grid pnts [K]-delzw: Delete Zero-weight gaussians from the GMM. ('T' or 'F') [T]-delid: Delete identical gaussians in the GMM. ('T' or 'F') [T]-nr: Number of repeat for EM [10000]-nk: Number of repeat for K-means multi-start [1]-cv: Convergence threshold for dParam for the EM algorithm [0.001000]-olog: Output logfile []-stdlog: output convergence log as stdout ('T' or 'F') [T]-ogmminit: Output Initial Gaussian File before EM algorithm (*.gmm) []-ogmmds: Output down-sampled Gaussian File for '-emalg D' or '-emalg DG'. (*.gmm) []-omapds: Output down-sampled CCP4 density map file (*.map) []-opost: Output PDB File for posterior probability []-justcnt: just count input number_of_atom /map_size, and quit. ('T' or 'F') [F]-rseed: random number seed(>=1) [1]
- Basic usage
gmconvert A2G -ipdb [pdb file] -ng [number of Gaussians] -ogmm [output GMM file]
- For example, if you want to convert the pdb file 'pdb5c44.ent' into GMM with 10 Gaussian functions, a command is as follows:
gmconvert A2G -ipdb pdb5c44.ent -ng 10 -ogmm 5c44_ng10.gmm
The generated GMM can be visually checked by various formats. The simplest way is just open the generated "*.gmm" file as the PDB file by molecular graphics program. Then, the centers of Gaussian functions are shown. - The isocontour surface model in VRML format (*.wrl) can be generated by the gmconvert by a following command:
gmconvert G2S -igmm 5c44_ng10.gmm -owrl 5c44_ng10_surface.wrl
- The ellipsoid model in VRML format (*.wrl) can be generated by the gmconvert by a following command:
gmconvert G2E -igmm 5c44_ng10.gmm -oewrl 5c44_ng10_ellipsoid.wrl
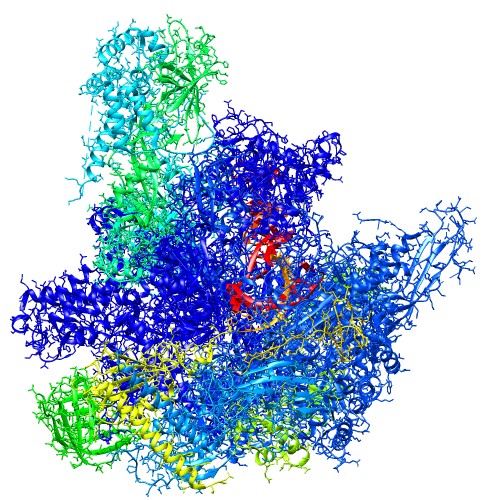



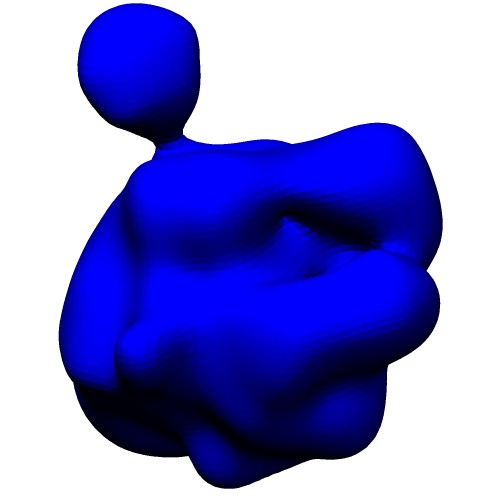
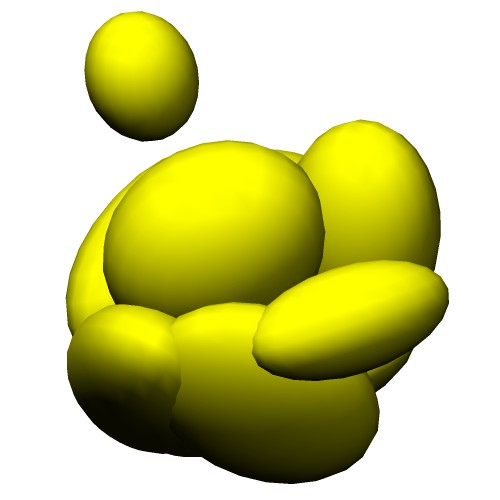
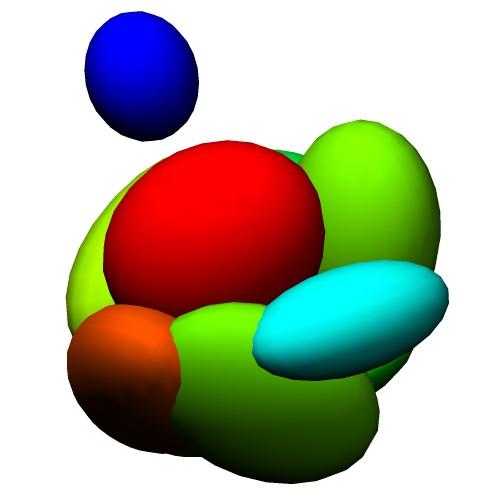
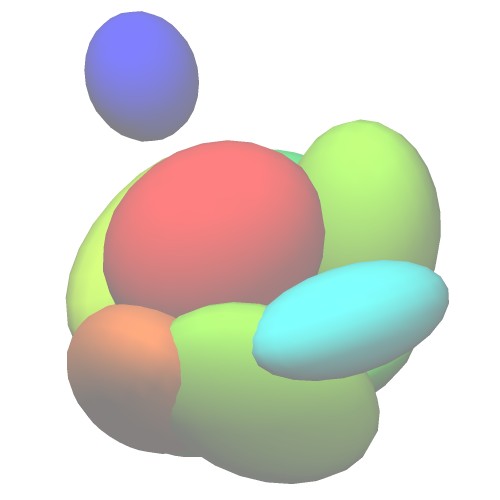
Atomic Model (PDB ID:5c44) Centers of GMM
(5c44_ng10.gmm)Wireframe of GMM isocontour surface
(5c44_ng10_surface.wrl)Ellipsoids of GMM
(5c44_ng10_ellipsoid.wrl) - Entire atoms are clustered into cubes with the edge of 20.0 angstrom, then each cube is converted into one Gaussian function.
gmconvert A2G -ipdb pdb5c44.ent -emalg D -dsatm E -wdsatm 20.0 -ogmm 5c44_D_Ew20.gmm
- Atoms in each chain is converted into one Gaussian function.
gmconvert A2G -ipdb pdb5c44.ent -emalg D -dsatm C -wdsatm 0.0 -ogmm 5c44_D_C.gmm
- Atoms in each secondary structure element is converted into one Gaussian function.
gmconvert A2G -ipdb pdb5c44.ent -emalg D -dsatm S -wdsatm 0.0 -ogmm 5c44_D_S.gmm
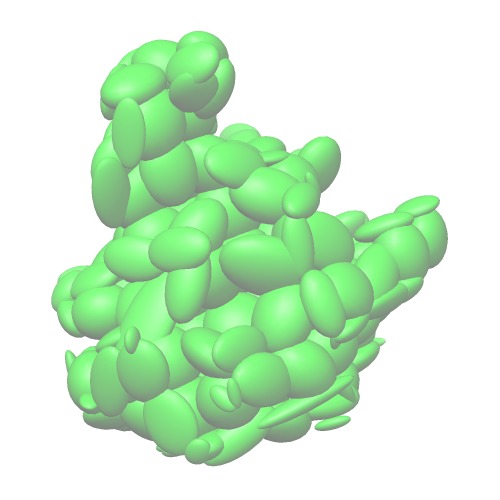


Down-sampledGMM.
Entire atoms with 20angstrom cubes
Ngauss=204(5c44_D_Ew20.gmm)Down-sampledGMM.
Each chain is converted into one Gaussian
Ngauss=15(5c44_D_C.gmm)Down-sampledGMM.
Each secondary structure is converted into one Gaussian
Ngauss=481(5c44_D_S.gmm) - Algorithms of fitting GMM with the atomic model
The program
gmconvertemploys the EM (expectarion-maximization) algorithm and down-sampling algorithm for fittting GMM with the given atomic model.
NOTE: The optionoption default or not name explanation -emalg PPoint-input GMM (P-input GMM) Each atom is represented by the center points with equal weights -emalg WPWeighted point-input GMM (WP-input GMM) Each atom is represented by the center points with atomic weights -emalg Gdefault Gaussian-input GMM (G-input GMM) Each atom is represented by the isotropic Gaussian function with variance = [Rvdw]/5. -emalg DDown-sampled GMM Atoms are clustered into groups, and atoms in a group are further divided into subgroups by PCA box. Each subgroup is represented by one Gaussian functin. -emalg DGdoes not work for the atomic model.-emalg D) quickly converts the atomic model into GMM. First, it clusters atoms into groups by 'E'ntire, 'C'hain, 'S'econdary structure, or 'R'esidue (with the option-dsatm). Secondly, atoms in each group are further clustered into subgroups using a PCA-box composed of cubes of width-wdsatm. Atoms in each subgroup are converted into one Gaussian function. If the widthwdsatm= 0, each group corresponds to one Gaussian function. - Options for input of the atomic model
The program gmconvert can read both PDB file and mmCIF file:
-ipdb: Input PDB file for atomic model []-icif: Input mmCIF file for atomic model []
You can restrict atoms in the PDB file as follows:
-hetatm: Read HETATM ('T' or 'F') [F].
The defaul is-hetatm F, it means the program only read "ATOM" line.-ch: Chain ID. (or 'auth_asym_id' in mmCIF).
The defaul is-ch -, it means the program reads all the chains in PDB file/mmCIF file.-atmsel: Atom selection. 'A'll atom except hydrogen, 'R'esidue-based (only ' CA ' and ' P ') [A]-model: 'S':read only single model (for NMR). 'M':read multiple models (for biological unit) [S]-IgnoreHydrogen: ignore hydrogen atoms and skip to read hydrogen atoms. ('T' or 'F')[T]
When a mmCIF file is input by the option
-icif, you can assign assembly_id for biological units:-assembly: assembly_id for mmCIF file (-icif) [].
Theassembly_idin mmCiF file (such as 1,2,3,PAU,XAU,..) can be assingned. The program performs symmetrc operations to asymmetric unit to generate XYZ coordinates of the assembly. If the optionassembly_idis not assigned, the program use the asymmetric unit.
- options for downsampling atom group
-dsatm: downsampling atom group for '-emalg D'. 'R'esidue,'S'ecstr, 'C'hain, 'M'odel 'E'ntire [E]-wdsatm: width of the cube of downsampling atom group [20.000000]-ndsatm: Division number for downsampling atom group. -wdsatm is tuned to make ng close to ndsatm [000] PCA division numbers can be assiged, such as '111','211','333'-owrldsatm: output VRML wire PCA box for downsampling atom group. []-opdbdsatm: output PDB file for downsampling atom group. []-rwdsatm: relative weight threshold for merging small Gauss into neighbors (0..1) [0.100000]
- Other options
-hetatm: Read HETATM ('T' or 'F') [F]-ch: Chain ID. (or 'auth_asym_id' in mmCIF). [-]-atmsel: Atom selection. 'A'll atom except hydrogen, 'R'esidue-based (only ' CA ' and ' P ') [A]-maxatm: maximum allowed number of atoms for '-atmsel A'. If over '-maxatm', then change '-atmsel R'.[-1]-atmrw: Model for radius and weight. 'A':atom model, 'R':residue model, 'M'ix of atom/residue. : 'U':uniform radius/weight,'C':decide from content. [C]-radtype: radius type for '-atmrw A'. V:van der Waals radius, C:covalent radius [V]-raduni: radius for uniform model for '-atmrw U'. [-1.000000]-varatm: Variance type for atom. 'R':sig = reso/2, 'A': sig^2= rr2var*Rvdw^2 + (reso/2)^2 'L': Laskowski style. f(Rvdw) = f(0.0)/2 'H':Hard-sphere-type [A]-rr2var: Constant for variance = Const * Rvdw*Rvdw for -emalg G -varatm A. Default is '1/5=0.2'. [0.200000].-reso: Resolution for atom-to-vox (sigma = reso/2) [0.000000]-orw: output radius-weight file []-uniRgmm: use uniform-Radius GMM ('T' or 'F') [F]-Runigmm: Radius for uniform-Radius GMM [3.800000]-ccatm: Calculation Corr Coeff bwn Atoms and GMMs.(It takes times..) (T or F)[F]-ogmmpc: Output Gaussian File with PC axis (*.gmm) []-ompdb: Output PDB File with membership values (*.pdb) []-impdb: Input PDB File with membership values (*.pdb) []-ogmm: Output Gaussian File (*.gmm) []-ng: Number of Gaussian functions for GMM [1]-emalg: type for EM algorithm. 'P'oint-input,'WP'eighted_point-input 'G'aussian-input (isotropic) 'O':1-to-1 atom/grid pnts : 'D':Down-sampled gaussians 'DG':Down-sampled gaussian-input GMM [G] : note: 'D' and 'DG' is only for map (V2G)-I: Initialization of GMM. 'K'-means, 'R'andom 'O':one-to-one_atom/grid pnts [K]-delzw: Delete Zero-weight gaussians from the GMM. ('T' or 'F') [T]-delid: Delete identical gaussians in the GMM. ('T' or 'F') [T]-nr: Number of repeat for EM [10000]-nk: Number of repeat for K-means multi-start [1]-cv: Convergence threshold for dParam for the EM algorithm [0.001000]-olog: Output logfile []-stdlog: output convergence log as stdout ('T' or 'F') [T]-ogmminit: Output Initial Gaussian File before EM algorithm (*.gmm) []-ogmmds: Output down-sampled Gaussian File for '-emalg D' or '-emalg d'. (*.gmm) []-omapds: Output down-sampled CCP4 density map file (*.map) []-opost: Output PDB File for posterior probability []-justcnt: just count input number_of_atom /map_size, and quit. ('T' or 'F') [F]-rseed: random number seed(>=1) [1]-max_memory: maximum memory size in mega byte [1000]