 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6qsw: Complement factor B protease domain in complex with the reversibl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6qsw | ||||||
|---|---|---|---|---|---|---|---|
| Title | Complement factor B protease domain in complex with the reversible inhibitor N-(2-bromo-4-methylnaphthalen-1-yl)-4,5-dihydro-1H-imidazol-2-amine. | ||||||
 Components Components | Complement factor B | ||||||
 Keywords Keywords | HYDROLASE / Complement / immune / inhibitor / c3 convertase | ||||||
| Function / homology |  Function and homology informationalternative-complement-pathway C3/C5 convertase / classical-complement-pathway C3/C5 convertase complex / complement binding / Alternative complement activation / Activation of C3 and C5 / complement activation, alternative pathway / complement activation / Regulation of Complement cascade / response to bacterium / blood microparticle ...alternative-complement-pathway C3/C5 convertase / classical-complement-pathway C3/C5 convertase complex / complement binding / Alternative complement activation / Activation of C3 and C5 / complement activation, alternative pathway / complement activation / Regulation of Complement cascade / response to bacterium / blood microparticle / serine-type endopeptidase activity / proteolysis / extracellular space / extracellular exosome / extracellular region / plasma membrane Function and homology informationalternative-complement-pathway C3/C5 convertase / classical-complement-pathway C3/C5 convertase complex / complement binding / Alternative complement activation / Activation of C3 and C5 / complement activation, alternative pathway / complement activation / Regulation of Complement cascade / response to bacterium / blood microparticle ...alternative-complement-pathway C3/C5 convertase / classical-complement-pathway C3/C5 convertase complex / complement binding / Alternative complement activation / Activation of C3 and C5 / complement activation, alternative pathway / complement activation / Regulation of Complement cascade / response to bacterium / blood microparticle / serine-type endopeptidase activity / proteolysis / extracellular space / extracellular exosome / extracellular region / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.64 Å | ||||||
 Authors Authors | Adams, C.M. / Sellner, H. / Ehara, T. / Mac Sweeney, A. / Crowley, M. / Anderson, K. / Karki, R. / Mainolfi, N. / Valeur, E. / Sirockin, F. ...Adams, C.M. / Sellner, H. / Ehara, T. / Mac Sweeney, A. / Crowley, M. / Anderson, K. / Karki, R. / Mainolfi, N. / Valeur, E. / Sirockin, F. / Gerhartz, B. / Erbel, P. / Hughes, N. / Smith, T.M. / Cumin, F. / Argikar, U. / Mogi, M. / Sedrani, R. / Wiesmann, C. / Jaffee, B. / Maibaum, J. / Flohr, S. / Harrison, R. / Eder, J. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2019 Title: Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Authors: Schubart, A. / Anderson, K. / Mainolfi, N. / Sellner, H. / Ehara, T. / Adams, C.M. / Mac Sweeney, A. / Liao, S.M. / Crowley, M. / Littlewood-Evans, A. / Sarret, S. / Wieczorek, G. / Perrot, ...Authors: Schubart, A. / Anderson, K. / Mainolfi, N. / Sellner, H. / Ehara, T. / Adams, C.M. / Mac Sweeney, A. / Liao, S.M. / Crowley, M. / Littlewood-Evans, A. / Sarret, S. / Wieczorek, G. / Perrot, L. / Dubost, V. / Flandre, T. / Zhang, Y. / Smith, R.J.H. / Risitano, A.M. / Karki, R.G. / Zhang, C. / Valeur, E. / Sirockin, F. / Gerhartz, B. / Erbel, P. / Hughes, N. / Smith, T.M. / Cumin, F. / Argikar, U.A. / Haraldsson, B. / Mogi, M. / Sedrani, R. / Wiesmann, C. / Jaffee, B. / Maibaum, J. / Flohr, S. / Harrison, R. / Eder, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6qsw.cif.gz | 590.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6qsw.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 6qsw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qs/6qswftp://data.pdbj.org/pub/pdb/validation_reports/qs/6qsw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6qsxC  6ravC  1dleS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| 3 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Components on special symmetry positions |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS ensembles :
|
-Components
| #1: Protein | / C3/C5 convertase / Glycine-rich beta glycoprotein / GBG / PBF2 / Properdin factor B Mass: 32955.883 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CFB, BF, BFD / Production host:  Drosophila melanogaster (fruit fly) Drosophila melanogaster (fruit fly)References: UniProt: P00751, alternative-complement-pathway C3/C5 convertase#2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 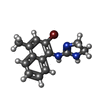  Mass: 304.185 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H14BrN3 Mass: 304.185 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C14H14BrN3#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 500 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 500 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 55.13 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 2.0 M ammonium sulfate, 0.1 M sodium acetate (pH 4.6), 2 mM inhibitor |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1.001 Å / Beamline: X10SA / Wavelength: 1.001 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Jun 1, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.001 Å / Relative weight: 1 |
| Reflection | Resolution: 1.64→65.1 Å / Num. obs: 129551 / % possible obs: 95.8 % / Redundancy: 7.6 % / Rrim(I) all: 0.068 / Net I/σ(I): 19.56 |
| Reflection shell | Resolution: 1.64→1.7 Å / Redundancy: 2.64 % / Mean I/σ(I) obs: 2.52 / Num. unique obs: 10441 / Rrim(I) all: 0.44 / % possible all: 76.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1DLE Resolution: 1.64→65.055 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.952 / SU B: 3.677 / SU ML: 0.054 / Cross valid method: FREE R-VALUE / ESU R: 0.089 / ESU R Free: 0.075 Details: Hydrogens have been added in their riding positions
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.87 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.64→65.055 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
|
